
Fetal alcohol spectrum disorder: molecular insights into neural damage reduction
2015-02-07 12:58DianaLeDuc
中國神經(jīng)再生研究(英文版) 2015年11期
Fetal alcohol spectrum disorder: molecular insights into neural damage reduction
Fetal alcohol spectrum disorders (FASD) is a group of entirely preventable, lifelong conditions, which occur upon maternal alcohol use during pregnancy. This can result in severe consequences for the newborn and ultimately the family. It is usually characterized by delays in development and motor function, craniofacial abnormalities, and diffi culties with learning, memory, speech, and academic achievement. According to the German guidelines for fetal alcohol syndrome (FAS) diagnosis, the prevalence of FASD ranges between 0.02–0.8% of all annual births and often the disorder is not recognized (Landgraf et al., 2013). The U.S. National Institutes of Health regard FAS as the most common nonhereditary cause of mental retardation. Thus, preventing programs, like the one undertaken by the Australian Government, which appointed a National FASD Technical Network (Elliott, 2015), may seem a very reasonable strategy. However, preventing programs for FASD focus mainly either on primary prevention, by increasing pregnant women’s awareness of ethanol consumption risks, or on tertiary prevention which supposes early recognition of the condition and social support in the form of an improved developmental framework of the aff ected individual. Secondary prevention of the disorder, which includes early detection by screening pregnant women for ethanol consumption and control of the progression from a preclinical pathological condition to a severe form of the disease, is considered to be most challenging (Elliott, 2015). Although the most important form of prevention is abstinence from alcohol during pregnancy, off springs from women with known gestational alcohol misuse could still benefi t from a secondary form of FASD prevention, which might hinder the progression to FAS. However, therapy approaches are precluded by our limited knowledge of molecular mechanisms responsible for neuronal damage.
Bosco and Diaz (2012) hypothesized that hypoxia and increased oxidative/nitrative stress are responsible for the decreased fetal growth, characteristic to FASD.
Under normal circumstances, during the transition from intra- to extrauterine life, newborns exhibit more or less pronounced levels of hypoxia, for which they are equipped with adaptive mechanisms that allow a striking high tolerance (Singer, 1999). We thus inquired whether alcohol exposure during the cerebellar growth spurt could lead to alterations of adaptive mechanisms to metabolic stressors (Le Duc et al., 2015).
To this end we exposed rats to 20% v/v ethanol in drinking water during the entire pregnancy period to induce ethanol exposure damage to the litter (Figure 1). For the rat neonatal cerebellum, the fi rst 10 days after birth correspond to the third pregnancy trimester in humans, when Purkinje cells grow dendrites and form synapses (Dobbing and Sands, 1979). Thus, in our study dams continued to receive ethanol 20% v/v in the drinking water and consequently off springs were exposed to ethanol through lactation until postnatal day 5, followed by an acute 3-hour ethanolvapor inhalation on postnatal day 5. Steady drinkers often show a behavioral pattern of chronic and acute binge ethanol consumption (Epstein et al., 1995), that was mimicked by our ethanol exposure model (Figure 1).
On postnatal day 5 cerebella were collected from ethanol exposed and control off springs and the granule neurons (CGNs) were cultured for 7 days in vitro (Figure 1). The first line of investigation focused on the morphology and in vitro adaptability of the neurons. No signifi cant diff erences were observed between the ethanol exposed and control CGNs for the 7 day period (Le Duc et al., 2015). This suggested that despite the long term, heavy ethanol exposure, CGNs retained similar adaptation capacities to an in vitro, nutrition rich environment as unexposed neurons. However, a direct ethanol exposure of CGN cultures causes a concentration-dependent depletion of the neurons (Pantazis et al., 1993) and in vivo ethanol exposure of the pups on postnatal day 7 leads to alteration in Rho GTPases signaling and increased apoptosis (Joshi et al., 2006). Taken together, the present lines of evidence could imply that while under ethanol exposure CGNs suff er extensive damage, but bringing previously exposed CGNs to normal conditions allows for their normal maturation.
Further, we investigated the impact of metabolic stressors on CGNs, which, despite the ethanol exposure during development, showed normal morphology in cell culture. Thus, after 7 days in vitro we exposed CGN cultures from ethanol-treated and control animals to either a serum free medium or to 3-hour oxygen glucose deprivation (OGD) (Figure 1). To assess the damage extent we performed dynamic propidium iodide measurements for 20 hours at 37°C in an exposure medium which contained glucose, but was serum free, mimicking nutritional metabolic stress.
Cellular death evaluation at the end of the 3-hour OGD revealed a signifi cantly higher damage in ethanol-treated neurons compared to control (7% vs. 0.65%). Maintenance of CGNs in normoxic/normoglycemic conditions resulted in a small but signifi cant diff erence in survival between the non-treated and the ethanol exposed groups (0.26% cell death vs. 0.62%). Thus, exposure of CGNs to ethanol during development causes a higher death susceptibility to an acute metabolic stress (Le Duc et al., 2015).
We then inquired the neuronal vulnerability after the 20 hour reoxygenation time. The eff ect of ethanol pre-exposure on CGNs’ susceptibility to OGD induced death was maintained over the 20 hour period of reoxygenation (33.4% cell death vs. 22.4% for the non-treated neurons).
To gain further insight into how ethanol pre exposure aff ects neuronal susceptibility to diff erent levels of metabolic defi cits we next focused on the dynamic recordings of neuronal delayed death over a 20 hour period (Figure 1). The low-nutrient propidium iodide containing experimental medium represented a metabolic stress, which induced a low, but constant rate of neuronal cell death in all CGN cultures. For the fi rst few hours of propidium iodide signal recording we observed no signifi cant diff erence between the ethanol pre-exposed neurons and the control ones. The sensitising eff ect of ethanol exposure became manifest between the cultures kept in normoxic/normoglycemic conditions only after 10–11 hours. The more damaging metabolic stress induced by OGD, resulted in a higher deathvulnerability of the ethanol treated neurons from the beginning of reoxygenation period (Le Duc et al., 2015). These results suggest that ethanol exposure of CGNs during development alters adaptive mechanisms to metabolic stressors, but the eff ect becomes apparent only after a highly damaging or prolonged metabolic insult. Such observations could be relevant for secondary prevention strategies of FASD, which aim to address the pre clinical pathological condition in order to hinder the progression to FAS. In our setup, despite the higher vulnerability of ethanol exposed neurons, increased cellular death happened only after specifi c triggers.
With a view to better understand the molecular mechanisms underlying FASD and given the observed eff ects of the diff erent stressors on the CGNs exposed to ethanol during development, we were further prompted to examine dysregulations in gene expression. Making use of a publicly available microarray dataset, in which whole embryo cultures were treated with ethanol to mimic FASD (Wang et al., 2008), we inquired Gene Ontology (GO) categories, which were aff ected by ethanol exposure. We focused specifi cally on fi ve categories which were signifi cantly aff ected in the ethanol exposed embryo cultures: oxidation-reduction process, mitochondrial respiratory chain, regulation of stress-activated MAPK cascade, in utero embryonic development, and nervous system development. The fi rst two showed an enrichment in genes which were down-regulated as a result of ethanol treatment, while the last three were enriched in upregulated genes. Using this approach we were able to defi ne candidate genes, which were further tested in our experimental model. The GO category “oxidation-reduction process” revealed an ethanol-induced up-regulation of Dhcr24 and a down-regulation of Cp and Snca. Dhcr24 was verifi ed by qPCR on the CGNs model, but Cp and Snca failed to reach significance. Additionally, the proapoptotic mitochondrial-membrane associated Bax was highly expressed in the ethanol group on CGNs, which could lead to apoptosis (Le Duc et al., 2015). However, under basal conditions we observed no change in the morphology of CGNs that could be indicative for increased apoptosis in the ethanol group. Overexpression of Dhcr24 was shown to inhibit apoptotic cell signalling (Lu et al., 2014), which could suggest an adaptive gene regulation to prevent the commitment on the activated apoptotic pathway in CGNs.
Bdnf was a gene that clustered in both the “oxidation-reduction process” and “nervous system development” Gene Ontology categories and appeared down-regulated following ethanol exposure of embryos. Bdnf acts protective on cerebellar granule neurons under low glucose conditions by preventing JNK and p38 activation (Vakili Zahir et al., 2012). On our in vitro model we measured an over expression of Mapk8 (Jnk1), which is known to promote apoptosis. Unlike on the whole embryo cultures Bdnf was up-regulated after ethanol exposure in our setup (Le Duc et al., 2015). It seems likely that up-regulation of Bdnf occurs as a counterbalance mechanism to maintain the viability of CGNs and we hence observe a higher vulnerability only under challenging conditions.
Based on our experimental data we propose that neurons exposed to ethanol during development show impaired adaptation to metabolic demanding circumstances. However, kept under normal conditions, there are compensatory mechanisms that allow for an apparent similar viability as that of normal developing neurons. This could imply that secondary prevention in FASD may be highly benefi cial by reducing the stress to which newborns from alcoholic mothers are exposed. Future research should address, preferably in in vivo studies whether reducing the mild hypoxia, which accompanies normal labour leads to a reduction in the brain damage of off springs with FASD and consequently a better long term prognostic.
Diana Le Duc*
Institute of Biochemistry, Molecular Biochemistry, Medical Faculty, University of Leipzig, Johannisallee 30, 04103 Leipzig, Germany
*Correspondence to: Diana Le Duc, Ph.D., Gabriela-Diana.LeDuc@medizin.uni-leipzig.de;
Diana_leduc@eva.mpg.de.
Accepted: 2015-07-31
Bosco C, Diaz E (2012) Placental hypoxia and foetal development versus alcohol exposure in pregnancy. Alcohol Alcohol 47:109-117.
Dobbing J, Sands J (1979) Comparative aspects of the brain growth spurt. Early Hum Dev 3:79-83.
Elliott EJ (2015) Fetal alcohol spectrum disorders in Australia--the future is prevention. Public Health Res Pract 25:e2521516.
Epstein EE, Kahler CW, McCrady BS, Lewis KD, Lewis S (1995) An empirical classifi cation of drinking patterns among alcoholics: binge, episodic, sporadic, and steady. Addict Behav 20:23-41.
Joshi S, Guleria RS, Pan J, Bayless KJ, Davis GE, Dipette D, Singh US (2006) Ethanol impairs Rho GTPase signaling and diff erentiation of cerebellar granule neurons in a rodent model of fetal alcohol syndrome. Cell Mol Life Sci 63:2859-2870.
Landgraf MN, Nothacker M, Heinen F (2013) Diagnosis of fetal alcohol syndrome (FAS): German guideline version 2013. Eur J Paediatr Neurol 17:437-446.
Le Duc D, Spataru A, Ceanga M, Zagrean L, Schoneberg T, Toescu EC, Zagrean AM (2015) Developmental exposure to ethanol increases the neuronal vulnerability to oxygen-glucose deprivation in cerebellar granule cell cultures. Brain Res 1614:1-13.
Lu X, Li Y, Wang W, Chen S, Liu T, Jia D, Quan X, Sun D, Chang AK, Gao B (2014) 3 beta-hydroxysteroid-Delta 24 reductase (DHCR24) protects neuronal cells from apoptotic cell death induced by endoplasmic reticulum (ER) stress. PLoS One 9:e86753.
Pantazis NJ, Dohrman DP, Goodlett CR, Cook RT, West JR (1993) Vulnerability of cerebellar granule cells to alcohol-induced cell death diminishes with time in culture. Alcohol Clin Exp Res 17:1014-1021.
Singer D (1999) Neonatal tolerance to hypoxia: a comparative-physiological approach. Comp Biochem Physiol A Mol Integr Physiol 123:221-234.
Vakili Zahir N, Abkhezr M, Khaje Piri Z, Ostad SN, Kebriaezade A, Ghahremani MH (2012) The time course of JNK and P38 activation in cerebellar granule neurons following glucose deprivation and BDNF treatment. Iran J Pharm Res 11:315-323.
Wang G, Wang X, Wang Y, Yang JY, Li L, Nephew KP, Edenberg HJ, Zhou FC, Liu Y (2008) Identifi cation of transcription factor and microRNA binding sites in responsible to fetal alcohol syndrome. BMC Genomics 9 Suppl 1:S19.
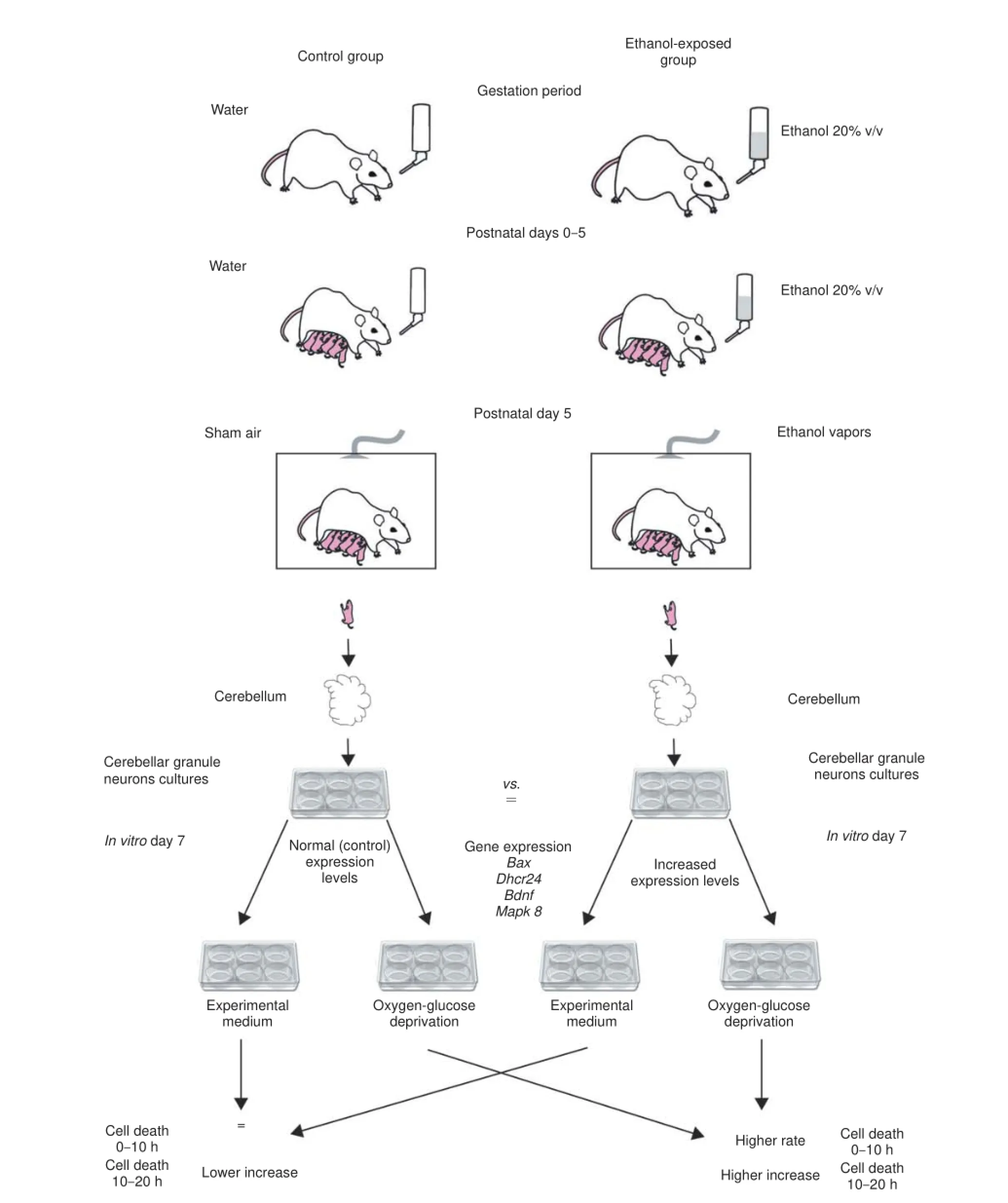
Figure 1 The impact of fetal ethanol exposure on metabolic stressors susceptibility.
10.4103/1673-5374.165290 http://www.nrronline.org/
Le Duc D (2015) Fetal alcohol spectrum disorder: molecular insights into neural damage reduction. Neural Regen Res 10(11):1764-1766.

- 中國神經(jīng)再生研究(英文版)的其它文章
- Intracellular sorting pathways of the amyloid precursor protein provide novel neuroprotective strategies
- The role of the Rho/ROCK signaling pathway in inhibiting axonal regeneration in the central nervous system
- VEGF in the nervous system: an important target for research in neurodevelopmental and regenerative medicine
- Studying neurological disorders using induced pluripotent stem cells and optogenetics
- Ef cacy of glucagon-like peptide-1 mimetics for neural regeneration
- Compliant semiconductor scaf olds: building blocks for advanced neural interfaces