
ARTICLE Excited-State Proton Transfer and Decay in Hydrogen-Bonded Oxazole System:MS-CASPT2//CASSCF Study?
2016-04-08 06:35BinbinXieChunxiangLiGanglongCuiQiuFangKeyLaboratoryofTheoreticalandComputationalPhotochemistryMinistryofEducationCollegeofChemistryBeijingNormalUniversityBeijing100875ChinaDatedReceivedonDecember2015AcceptedonDecember
Bin-bin Xie,Chun-xiang Li,Gang-long Cui?,Qiu Fang?Key Laboratory of Theoretical and Computational Photochemistry,Ministry of Education,College of Chemistry,Beijing Normal University,Beijing 100875,China(Dated:Received on December 1,2015;Accepted on December 30,2015)
?
ARTICLE Excited-State Proton Transfer and Decay in Hydrogen-Bonded Oxazole System:MS-CASPT2//CASSCF Study?
Bin-bin Xie,Chun-xiang Li,Gang-long Cui?,Qiu Fang?
Key Laboratory of Theoretical and Computational Photochemistry,Ministry of Education,College of Chemistry,Beijing Normal University,Beijing 100875,China
(Dated:Received on December 1,2015;Accepted on December 30,2015)
Herein we have employed high-level multi-reference CASSCF and MS-CASPT2 electronic structure methods to systematically study the photochemical mechanism of intramolecularly hydrogen-bonded 2-(2′-hydroxyphenyl)-4-methyloxazole.At the CASSCF level,we have optimized minima,conical intersections,minimum-energy reaction paths relevant to the excited-state intramolecular proton transfer(ESIPT),rotation,photoisomerization,and the excited-state deactivation pathways.The energies of all structures and paths are re fi ned by the MS-CASPT2 method.On the basis of the present results,we found that the ESIPT process in a conformer with the OH···N hydrogen bond is essentially barrierless process; whereas,the ESIPT process is inhibited in the other conformer with the OH···O hydrogen bond.The central single-bond rotation of the S1enol species is energetically unfavorable due to a large barrier.In addition,the excited-state deactivation of the S1keto species,as a result of the ultrafast ESIPT,is very e ffi cient because of the existence of two easily-approached keto S1/S0conical intersections.In stark contrast to the S1keto species,the decay of the S1enol species is almostly blocked.The present theoretical study contributes valuable knowledge to the understanding of photochemistry of similar intramolecularly hydrogen-bonded molecular and biological systems.
Keywords:Excited state proton transfer,Photoisomerization,Conical intersection,Ab initio,Photochemistry
?Part of the special issue for“the Chinese Chemical Society’s 14th National Chemical Dynamics Symposium”.
?Authors to whom correspondence should be addressed.E-mail: ganglong.cui@bnu.edu.cn,fangqiu917@bnu.edu.cn
I.INTRODUCTION
Excited state intramolecular proton transfer(ESIPT) and its subsequent photodynamics play an important role in a lot of biological processes[1?8]and in numerous applications such as photostabilizers[9]UV fi lter materials[10?12], fl uorescent probes[13],and sunscreens[14].Due to its importance,this kind of photochemical reactions has been extensively studied by experimental and theoretical chemists in past decades [15?37].

FIG.1 2-(2'-Hydroxyphenyl)-4-methyloxazole molecule in which there is a strong intramolecular hydrogen bond O?H···Nenablingexcited-stateintramolecularproton transfer between the enol and keto conformers.Also shown are the competitive single-bond rotation(left)and doublebond photoisomerization(right)channels.
In this work,we focus on the system of 2-(2′-hydroxyphenyl)-4-methyloxazole(HPMO),as shown in Fig.1.Experimental study of excited-state dynamics of HPMO can be dated back to the end of the last century.Guallar et al.experimentally studied the ESIPT and rotational processes of 2-(2′-hydroxyphenyl)-oxazole derivatives including HPMO in both S0and S1states and supported the coexistence of two groundstate conformers with OH···N and OH···O hydrogen bonds[38].Interestingly,only a conformer was observed to experience a photoinduced proton transfer.Zewail et al.studied the femtosecond dynamics of HPMO in con fi ned nanocavities and in aprotic solvents[39].They suggested that the ESIPT process occurs within 300 fs in aprotic solvents;whereas,in con fi ned nanocavities, this process is slowed down to a subpicosecond time scale.In addition,they also found a picosecond twisting motion around the central single bond,which is noticeably inhibited inside the nanocavities.Garc′?a-Ochoa et al.explored the ESIPT process of HPMO in various hydrophobic nanocavities in aqueous medium[40].In their experiments,upon irradiation,a fast ESIPT re-action produces a phototautomer with a large Stokes shift.Furthermore,they also found a twisting motion around the central single bond of this generated phototautomer.Later,Zhong et al.further explored the femtosecond dynamics of HPMO in human serum albumin protein,also in micelles and cyclodextrins for comparison[41].They found that the con fi ned geometry restrains the nonradiative decay and thus significantly extends the excited-state lifetime.Their most important fi nding is that the ESIPT and subsequent intramolecular twisting proceed in two di ff erent routes. The fi rst is the direct in-plane stretching motion,about 200 fs,which is insensitive to the surroundings.The second is less dominant and is related to the out-ofplane twisting motion(ca.3 ps)of the two heterocyclic rings,which is drastically slowed down in the protein hydrophobic environment.
On the theoretical side,there exist merely a few crude theoretical calculations at the semiempirical,Hartree-Fock(HF)and con fi guration interaction with single excitation(CIS)levels.Douhal et al.employed the HF and CIS methods to study the ESIPT processes in the S0and S1states,respectively[38].Guallar and coworkers performed semiclassical molecular dynamics simulations for the ESIPT process,which is however based on the CIS computed potential energy surface [42].Lluch et al.also studied the ESIPT process of HPMO embedded in β-cyclodextrin using the HF and CIS-based ONIOM methods[43,44].Hamms-Schi ff er et al.simulated the ultrafast ESIPT process of HPMO in vacuo,solution,and protein environments using classical molecular dynamics in conjunction with an empirical valence bond potential[45].They found that the ring-ring bending motion is the most important low-frequency vibrational mode,which helps decrease the proton-acceptor distance and thus facilitates proton transfer;the S1decay is much slower in water than in aprotic solvents and protein,which is ascribed to the fact that intermolecular hydrogen-bonding leads to a disruption of the intramolecular hydrogen-bonding in HPMO.
However,previous theoretical studies only focus on the ESIPT process of the excited-state dynamics of HPMO;thus,a few essential mechanistic details remain unknown,for example,how does the generated phototautomer decay to the S0state?Furthermore,it is well known that excited-state deactivation is usually related to conical intersections.Near these quasi-degenerate regions,multi-reference electronic structure methods must be used to get a correct description of topological structures of relevant potential energy surfaces.Herein, we have for the fi rst time employed the high-level complete active space self-consistent fi eld(CASSCF)and its multi-state second-order perturbation theory(MSCASPT2)methods to study the ESIPT and rotational processes,and the S1excited-state deactivation channels.
II.COMPUTATIONAL DETAILS
Minima(S0and S1),minimum-energy conical intersections(MECI,S1/S0),and minimum-energy reaction paths(S0and S1)are computed using the stateaveraged complete active space self-consistent fi eld(SACASSCF)method in which equal state weights are used for both electronic states.In all SA-CASSCF geometric optimizations,an active space of 10 electrons in 8 orbitals is used,which includes 10π electrons in 8π and π?orbitals(Fig.2).To obtain more accurate potential energy pro fi les,the MS-CASPT2 method[46, 47]that provides more correlation energy is exploited to re-evaluate the energies of all CASSCF optimized geometries and reaction paths.In single-point MSCASPT2 calculations,an imaginary shift of 0.2 a.u.is used to avoid the intruder-state issue[48];the Cholesky decomposition technique with unbiased auxiliary basis sets is used for accurate two-electron integral approximations[49];the ionization potential-electron a ffi nity (IPEA)shift was not applied[50].This combined MSCASPT2//CASSCF computational strategy enables a good description for photophysics and photochemistry of medium-size molecular systems in vacuo,solution, and proteins,as demonstrated in many our previous computational studies[16,51?59].
Vertical excitation energies are computed using TD-CAM-B3LYP[60],TD-B3LYP[61?64],and MSCASPT2 methods,respectively.The 6-31G?basis set [65,66]is used for all computations.All TD-DFT computations and CASSCF optimizations of conical intersections are carried out using Gaussian 09[67];all other CASSCF computations and MS-CASPT2 computations are performed using MOLCAS 8.0[68].
III.RESULTS AND DISCUSSION
Figure 3 shows the schematic structures optimized at the CASSCF(10,8)/6-31G?level.Table I lists the selected geometric parameters and the MS-CASPT2 ref
i ned energies. A.S0minima and vertical excitation energies
At the CASSCF level,we have obtained three S0conformers,which are denoted as S0-ENOL-1,S0-KETO, and S0-ENOL-2,respectively.Of them,S0-ENOL-1 and S0-ENOL-2 are the most stable two conformers at this computational level;while,S0-KETO is 18.7 and 13.2 kcal/mol higher than S0-ENOL-1 and S0-ENOL-2 in energy(Table I).

FIG.2 Eight active orbitals in the CASSCF(10,8)/6-31G*computations.

FIG.3 CASSAF(10,8)/6-31G?optimized S0and S1minima(bond length in?A).See supplementay material for their Cartesian coordinates.Table I collects their relative energies re fi ned by the MS-CASPT2 method.

TABLE I Selected geometric parameters(CASSCF level, bond angles and dihedral angles in(?))and MS-CASPT2 re fi ned energies E(in kcal/mol).
The vertical excitation energy to the fi rst excited single state S1at the enol Franck-Condon point of HPMO shows that this S0→S1vertical excitation energy is computed to be 4.2 eV at the MS-CASPT2 level and TD-B3LYP level,which is about 0.2 eV lower than that computed by the TD-CAM-B3LYP method (4.4 eV)and is about 0.3 eV higher than the experimental value measured in solution[41].We have analyzed the molecular orbitals relevant to the S0→S1 electronic transition of the enol minimum S0-ENOL-1, as shown in Fig.2.The S1state is a spectroscopically bright state being ππ?character.At the CASSCF level,there are two main transition components for the S0→S1electronic transition.One is from HOMO?2 to LUMO(weight:0.317)and another from HOMO?1 to LUMO+1(0.183).Accordingly,there are four activespace orbitals whose electronic occupations signi fi cantly deviate from empty or full one.It can also be found that HOMO?2 and LUMO+1 are localized within the left six-membered group;whereas,HOMO?1 and LUMO spread over the whole molecular space.Thus,we can observe partial electron transfer from the phenyl group (HOMO?2)to the methyloxazole group(LUMO)in the S0→S1electronic transition.
B.S1excited-state minima
In addition,we have optimized three S1minima at the CASSCF level,which are denoted as S1-ENOL-1, S1-KETO and S1-ENOL-2.According to the adiabatic excitation energies collected in Table I,it is clear thatat the MS-CASPT2 level,S1-ENOL-1 is 2.3 kcal/mol higher than S1-KETO and 7.4 kcal/mol lower than S1-ENOL-2,respectively;S1-KETO is 9.9 kcal/mol lower than S1-ENOL-2.

FIG.4 Four molecular orbitals whose electronic occupations signi fi cantly deviate from empty(0.0)or full(2.0)occupation involved in the S0→S1electronic transition at the enol S1minimum.

FIG.5 Schematic S1/S0conical intersections S1S0-1(left),S1S0-2(middle),and S1S0-3(right).Also shown are their two singly-occupied molecular orbitals and selected bond lengths in?A.
As shown in Fig.3,the N1?H6 bond length of S1-ENOL-1 is decreased to 1.80?A from 1.91?A of S0-ENOL-1,which is a clear evidence that the excitedstate hydrogen-bonding interaction is reinforced in the S1state.The C2?C3 bond length of S1-ENOL-1 is also strengthened,which is about 0.04?A shorter than that of S0-ENOL-1.The similar changes are seen for S0-ENOL-2 and S1-ENOL-2.At S1-KETO,the H6 has already transferred to the N1 atom;the O5?H6 bond is increased by 0.12?A relative to that of S0-KETO,which implies the N1···H6 hydrogen bond is weakened.
C.S1/S0conical intersections
We have optimized three S1/S0conical intersections at the CASSCF level,which are denoted as S1S0-1, S1S0-2 and S1S0-3.S1S0-1 and S1S0-2 are structurally almost equivalent(Fig.5).They are located in the keto region i.e.after the H6 atom transferred to the N1 atom.Structurally,we can fi nd a strong pyramidalization at one C atom after the twisting.This could originate from the sudden polarization e ff ects,as seen in many similar systems[69?71].Table I shows that the energies of S1S0-1 and S1S0-2 are very close to each other,which are computed to be 80.3 and 82.3 kcal/mol at MS-CASPT2 level,respectively.By contrast,S1S0-3 corresponds to a conical intersection with the broken C2?O7 bond.Its energy is also close to the other two conical intersections within about 1 kcal/mol at the MS-CASPT2 level.Finally,we should note that at MS-CASPT2 level,all these three conical intersections are energetically allowed if only considering their energies relative to the S1energy at the enol Franck-Condon point i.e.S0-ENOL-1,which is about 95.7 and 101.5 kcal/mol at MS-CASPT2 and TD-CAM-B3LYP levels,respectively.However,their importance in the photodynamics of HPMO is very distinct(vide infra).
D.Excited-state rotation
Does the central C?C bond rotation take place easily?The answer is not.At the MS-CASPT2 level,we have computed the S1minimum-energy rotational path of HPMO.As shown in Fig.6,it is transparent that the S1barrier for the rotation from S1-ENOL-1 to S1-ENOL-2 is more than 20 kcal/mol,which is much higher than the counterpart in the S0state.Clearly,this process cannot compete with the in-plane S1excited-state intramolecular proton transfer.
E.Excited-state proton transfer
There are two types of S1excited-state intramolecular proton transfer in HPMO.The fi rst is from the O atom of the six-membered ring to the N atom of the fi ve-membered ring,which is barrierless and thus effi cient;whereas,the second,from the O atom of the six-membered ring to the O atom of the fi ve-membered ring,becomes inhibited due to a much higher barrier.
The fi rst S1excited-state intramolecular proton transfer starts from the spectroscopically bright S1state that is of ππ?character at the enol minimum S0-ENOL-1.Upon excitation to this1ππ?state at the enol Franck Condon point,the system fi rst arrives at a shallow S1minimum referred to as S1-ENOL-1 in Fig.3. At this st?ructure,the N?1···H6 bond length is decreasedto 1.80A from 1.91A of the S0enol minimum S0-ENOL-1,which is a clear evidence that the hydrogen bond is reinforced in the S1(1ππ?)state.This kind of enhancement is also seen in our recent several theoretical work on excited-state intramolecular proton transfers[36,72].This hydrogen-bond shortening bene fi ts the subsequent S1excited-state intramolecular proton transfer.From the S1enol minimum S1-ENOL-1,an ultrafast excited-state proton transfer could be expected, forming an S1keto minimum S1-KETO.This point of view is supported by the MS-CASPT2//CASSCF computed S1minimum-energy proton transfer path in Fig.7. The S1potential energy surface with respect to the N1?H6 bond length is very fl at and essentially barrierless(0.7 kcal/mol at the MS-CASPT2 level).In addition,we have found that the driving force for this S1ESIPT process is not so strong because the reaction energy change is only within several kcal/mol at the MS-CASPT2 level.Thus,there should exist an equilibrium between the S1enol and keto minima.This kind of S1excited-state intramolecular proton transfer induced equilibrium is rarely reported computationally. In most of our previous computational studies,the S1excited-state intramolecular proton transfer usually corresponds to a much exothermic process[72?74].

FIG.7 MS-CASPT2//CASSCF computed S1minimumenergy proton-transfer path(relaxed1ππ?state).

FIG.8 MS-CASPT2//CASSCF computed S1minimumenergy proton-transfer path(relaxed1ππ?state).
The second S1excited-state intramolecular proton transfer starts from another S1enol minimum S1-ENOL-2.It is clear that this process is thermodynamically unfavorable in the S1state at the MS-CASPT2 level in that the S1energy increases with the increasing O7?H6 bond length(Fig.8).Considering that it is also very di ffi cult for HPMO to transform from S1-ENOL-1 to S1-ENOL-2 in Fig.6(more than 20 kcal/mol at MS-CASPT2),it is safe to expect that this latter S1excited-state intramolecular proton transfer is entirely blocked in the photodynamics of HPMO.

FIG.9 MS-CASPT2//CASSCF computed S1minimumenergy reaction path with regard to the O7?C2 bond length. It connects the enol1ππ?minimum S1-ENOL-1 and the enol minimum-energy S1/S0conical intersection S1S0-3.
F.Deactivation path of the S1enol species
In addition to the ultrafast,barrierless S1excitedstate intramolecular proton transfer as mentioned above,the S1enol minimum S1-ENOL-1 can also undergo an S1excited-state decay via the S1/S0conical intersection with the broken C?O bond i.e.S1S0-3 (see Fig.3).However,this S1excited-state deactivation channel is nearly blocked because its related S1barrier, on the basis of the MS-CASPT2//CASSCF computed S1minimum-energy reaction path in Fig.9,is predicted to be 21.9 kcal/mol,which cannot be overcome concerning the S1energy of HPMO at the enol Franck-Condon point.
G.Deactivation path of the S1keto species
In contrast to the S1enol species,there exist e ffi cient S1excited-state decay pathways connecting the S1keto species and the keto S1/S0conical intersections S1S0-1 and S1S0-2.At the MS-CASPT2//CASSCF level,we have computed the corresponding S1minimum-energy reaction path along the rotation of the N1?C2?C3?C4 dihedral angle,which is shown in Fig.10.It is clear there are two quasi-degenerate regions,which are located at the positions with the dihedral angle of 60?and 130?, respectively.In fact,these two regions are close to the two keto S1/S0conical intersections S1S0-1 and S1S0-2. As mentioned before,these two conical intersections are energetically allowed because their energies are all lower than the S1energy at the enol Franck-Condon point.
Next,we will show they can also be accessed from their nearby S1keto species.Apparently,it is very easy for the S1keto species to arrive at the fi rst keto S1/S0conical intersection i.e.S1S0-1 because there only exists a small barrier of 3.7 kcal/mol at the MS-CASPT2 level(see Fig.10,at about 60?).At this hopping area, the S1system can be de-excited to the S0state and then recover to its initial enol S0minima S0-ENOL-1 or S0-ENOL-2.Importantly,if the system does not hop to the S0state when it encounters the fi rst keto S1/S0conical intersection S1S0-1,the S1keto species still can decay to the S0state at the second keto S1/S0conical intersection S1S0-2.Taking these two aspects in account,we can conclude that the excited-state deactivation starting from the S1keto species is very e ffi cient and could be an ultrafast process.
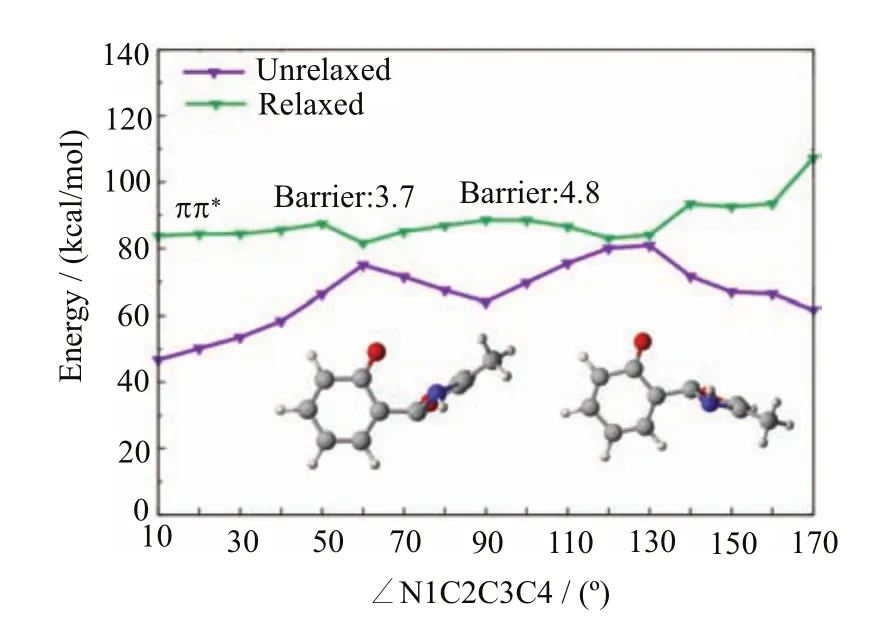
FIG.10 MS-CASPT2//CASSCF computed S1minimumenergy reaction path along the rotation of the N1-C2-C3-C4 dihedral angle connecting the keto1ππ?minimum S1-KETO and the two keto minimum-energy S1/S0conical intersections S1S0-1 and S1S0-2.
H.Mechanism
On the basis of the present results,we can summarize the photophysical and photochemical mechanism of HPMO in Fig.11.Upon irradiation to the bright S1state at the enol Franck-Condon point,the system fi rst relaxes to a nearby local S1minimum,which is referred to as S1-ENOL-1 in Fig.3.Starting from this point, there exist two competitive S1relaxation channels.The fi rst one is the nearly barrierless S1excited state intramolecular proton transfer from the O atom of the six-membered ring to the N atom of the fi ve-membered ring.Its related barrier is estimated to be 0.7 kcal/mol at the MS-CASPT2 level.This ultrafast process generates a planar S1keto species,which should be able to fl uoresce in rigid surroundings because steric interaction can signi fi cantly prevent the central C?C bond rotation.Instead,in vacuo or in low-viscosity solution, the C?C bond rotation becomes rather easy,which only needs to overcome a small barrier of 3.7 kcal/mol at the MS-CASPT2 level.Mechanistically,this facile rotation induces an e ffi cient excited-state deactivation via the two keto S1/S0conical intersections S1S0-1 and S1S0-2,which are located near the rotational pathway of the central C?C bond.On hopping to the S0state, the vibrationally“hot”molecule can move to the two enol S0minima,either S0-ENOL-1 or S0-ENOL-2.In the second one,the enol S1species can decay to the S0state via the enol S1/S0conical intersection S1S0-3.However,this relaxation channel is completely prohibited due to the existing large barrier,which is about 21.9 kcal/mol at the MS-CASPT2 level,even higher than the S1energy at the enol Franck-Condon point S0-ENOL-1,95.7 and 101.5 kcal/mol at MS-CASPT2 and TD-CAM-B3LYP levels,respectively.In addition,this process also cannot compete with the essentially barrierless S1excited-state intramolecular proton transfer. Considering these factors,this second decay pathway is mechanistically unimportant.Figure 11 schematically shows our suggested photochemical mechanism based on the present theoretical study.

FIG.11 Photophysical and photochemical mechanism of HPMO suggested based on the present MS-CASPT2//CASSCF electronic structure calculations.Relative energies are also shown(kcal/mol).
IV.CORRELATION WITH PREVIOUS WORK
Our proposed photochemical mechanism rationalizes the phenomena of experiments available.We found that the ESIPT process happens only for S1-ENOL-1, which explains very well the observation of Guallar et al.[38]and Zewail et al.[39,41].In their experiments, only a conformer was observed to experience a photoinduced proton transfer and the ESIPT process occurs within subpicosecond in aprotic solvents and con fi ned nanocavities.In addition,the generated S1keto species can twist its central C?C bond to arrive at the S1/S0conical intersection so as to decay to the ground state. This process is demonstrated to be e ffi cient owing to a small barrier of ca.3 kcal/mol at the MS-CASPT2 level.This also rationalizes why previous experiments found a picosecond twisting motion around the central single bond of the phototautomer[41].Since the rotational motion involves a large conformation change,it must be noticeably inhibited inside the nanocavities due to steric interaction.This fi ts very well with the conclusion of Zewail and coworkers:“the con fi ned geometry restrains the nonradiative decay and thus signi fi cantly extends the excited-state lifetime”[41].
Furthermore,our work provides new mechanistic insights.First,correct and accurate potential energy pro fi les are attained,which plays a key role in understanding the photochemical mechanism of HPMO and its derivatives.At the CIS level,Douhal et al. predicted the S1barrier related to ESIPT is more than 10 kcal/mol for 2-(2′-hydroxyphenyl)-4-oxazole [38].Due to the use of single-reference methods in previous theoretical works,the potential energy pro fi les close to the S1/S0conical intersections,for example those related to the excited-state decay of the S1keto species, are incorrectly described.For instance,Lluch et al. predicted a barrier of ca.8 kcal/mol for the central C?C bond rotation of the S1keto species in isolated HPMO and HPMO/β-CD complex[44].Instead,both S1and S0states should be close to each other along this rotational motion,as shown in Fig.10.Second,we have located several enol and keto S1/S0conical intersections and their S1deactivation channels,which is helpful for understanding the nonradiative dynamics of HPMO and its variants.
V.CONCLUSION
By means of high-level CASSCF and MS-CASPT2 methods,we have systematically explored the photophysical and photochemical mechanism of HPMO. The S1and S0minima,S1/S0MECIs,and minimumenergy reaction paths relevant to the S1excited-state intramolecular proton transfer and the S1enol and keto species decay channels are optimized at the CASSCF level and re fi ned at the MS-CASPT2 level.In terms of the present results,we fi nd that the excited-state intramolecular proton transfer is an overwhelmingly dominant relaxation pathway for the S1enol species and is expected to be an ultrafast process.It completely defeats the S1excited-state decay via the enolS1/S0MECI with a large barrier.The produced S1keto species should be able to fl uoresce if its central C?C bond rotation is inhibited in certain rigid surroundings,such as in solid states or high-viscosity solution.On the contrary,this S1keto species will decay to the S0state in an ultrafast means via the two keto S1/S0MECIs that can be easily approached in vacuo and dilute solution.Then,the S0enol minima are re-populated again.The present high-level electronic structure calculations provide many valuable mechanistic insights and could help understand the photodynamics of HPMO and other similar intramolecularly hydrogen-bonded molecular systems.
Supplementary materials:Cartesian coordinates of all optimized structures are shown.
VI.ACKNOWLEDGMENTS
This work was supported by the National NaturalScienceFoundationofChina(No.21522302, No.21520102005,and No.21421003).Gang-long Cui is also grateful for fi nancial support from the Recruitment Program of Global Youth Experts Youth Scholars Program of Beijing Normal University,Fundamental Research Funds for Central Universities.
[1]P.F.Barbara,P.K.Walsh,and L.E.Brus,J.Phys. Chem.93,29(1989).
[2]W.E.Brewer,M.L.Martnez,and P.T.Chou,J.Phys. Chem.94,1915(1990).
[3]T.Arthen-Engeland,T.Bultmann,N.P.Ernsting,M. A.Rodriguez,and W.Thiel,Chem.Phys.163,43 (1992).
[4]A.Sytnik and M.Kasha,Proc.Natl.Acad.Sci.91, 8627(1994).
[5]T.Mutai,H.Tomoda,T.Ohkawa,Y.Yabe,and K. Araki,Angew.Chem.Int.Ed.47,9522(2008).
[6]F.A.S.Chipem and G.Krishnamoorthy,J.Phys. Chem.A 113,12063(2009).
[7]L.Antonov,V.Deneva,S.Simeonov,V.Kurteva,D. Nedeltcheva,and J.Wirz,Angew.Chem.Int.Ed.48, 7875(2009).
[8]S.Park,J.E.Kwon,and S.Y.Park,Phys.Chem. Chem.Phys.14,8878(2012).
[9]M.J.Paterson,M.A.Robb,L.Blancafort,and A.D. DeBellis,J.Phys.Chem.A 109,7527(2005).
[10]S.Park,O.H.Kwon,S.Kim,S.Park,M.G.Choi,M. Cha,S.Y.Park,and D.J.Jang,J.Am.Chem.Soc. 127,10070(2005).
[11]J.E.Kwon,S.Park,and S.Y.Park,J.Am.Chem. Soc.135,11239(2013).
[12]W.Zhang,Y.L.Yan,J.M.Gu,J.N.Yao,and Y.S. Zhao,Angew.Chem.Int.Ed.54,7125(2015).
[13]B.Gu,L.Y.Huang,N.X.Mi,P.Yin,Y.Y.Zhang,X. M.Tu,X.B.Luo,S.L.Luo,and S.Z.Yao,Analyst 140,2778(2015).
[14]M.T.Ignasiak,C.Hou′ee-Levin,G.Kciuk,B. Marciniak,and T.Pedzinski,ChemPhysChem 16,628 (2015).
[15]G.Yang,F.Morlet-Savary,Z.Peng,S.Wu,and J.P. Fouassier,Chem.Phys.Lett.256,536(1996).
[16]W.H.Fang,J.Am.Chem.Soc.120,7568(1998).
[17]A.L.Sobolewski and W.Domcke,Phys.Chem.Chem. Phys.1,3065(1999).
[18]S.Lochbrunner,A.J.Wurzer,and E.Riedle,J.Chem. Phys.112,10699(2000).
[19]A.L.Sobolewski and W.Domcke,J.Phys.Chem.A 108,10917(2004).
[20]M.Zi′o lek,J.Kubicki,A.Maciejewski,R.Naskr?ecki, and A.Grabowska,Phys.Chem.Chem.Phys.6,4682 (2004).
[21]D.Nedeltcheva,B.Damyanova,and S.Popov,J.Mol. Struct.749,36(2005).
[22]Y.Wu and V.S.Batista,J.Chem.Phys.124,224305 (2006).
[23]A.Sobolewski and W.Domcke,J.Phys.Chem.A 111, 11725(2007).
[24]A.Migani,M.Bearpark,M.Olivucci,and M.Robb,J. Am.Chem.Soc.129,3703(2007).
[25]W.Rodr′?guez-C′ordoba,J.S.Zugazagoitia,E.Collado-Fregoso,and J.Peon,J.Phys.Chem.A 111,6241 (2007).
[26]A.Migani,L.Blancafort,M.A.Robb,and A.D.De-Bellis,J.Am.Chem.Soc.130,6932(2008).
[27]G.J.Zhao and K.L.Han,Phys.Chem.Chem.Phys. 12,8914(2010).
[28]K.C.Tang,M.Chang,T.Y.Lin,H.A.Pan,T.C. Fang,K.Y.Chen,W.Y.Hung,Y.H.Hsu,and P.T. Chou,J.Am.Chem.Soc.133,17738(2011).
[29]G.J.Zhao and K.L.Han,Acc.Chem.Res.45,404 (2012).
[30]G.L.Cui and W.Thiel,Phys.Chem.Chem.Phys.14, 12378(2012).
[31]T.Sekikawa,O.Schalk,G.Wu,A.E.Boguslavskiy,and A.Stolow,J.Phys.Chem.A 117,2971(2013).
[32]N.Suzuki,A.Fukazawa,K.Nagura,S.Saito,H.Kitoh-Nishioka,D.Yokogawa,S.Irle,and S.Yamaguchi, Angew.Chem.Int.Ed.53,8231(2014).
[33]D.Tuna,A.Sobolewski,and W.Domcke,J.Phys. Chem.B 118,976(2014).
[34]X.P.Chang,Q.Fang,and G.L.Cui,J.Chem.Phys. 141,154311(2014).
[35]S.H.Xia,B.B.Xie,Q.Fang,G.L.Cui,and W.Thiel, Phys.Chem.Chem.Phys.17,9687(2015).
[36]P.J.Guan,G.L.Cui,and Q.Fang,ChemPhysChem 16,805(2015).
[37]X.P.Chang,G.L.Cui,W.H.Fang,and W.Thiel, ChemPhysChem 16,933(2015).
[38]V.Guallar,M.Moreno,J.M.Lluch,F.Amat-Guerri, and A.Douhal,J.Phys.Chem.100,19789(1996).
[39]A.Douhal,T.Fiebig,M.Chachisvilis,and A.H.Zewail, J.Phys.Chem.A 102,1657(1998).
[40]I.Garc′?a-Ochoa,M.A.D.L′opez,M.H.Vi?nas,L.Santos,E.M.At′az,F.Amat-Guerri,and A.Douhal,Chem. Eur.J.5,897(1999).
[41]D.P.Zhong,A.Douhal,and A.H.Zewail,Proc.Natl. Acad.Sci.USA 97,14056(2000).
[42]V.Guallar,V.S.Batista,and W.H.Miller,J.Chem. Phys.113,9510(2000).
[43]R.Casades′us,M.Moreno,and J.M.Lluch,Chem. Phys.Lett.356,423(2002).
[44]R.Casades′us,M.Moreno,and J.M.Lluch,Photobiol. 173,365(2005).
[45]O.Vendrell,M.Moreno,J.M.Lluch,and S.Hammes-Schi ff er,J.Phys.Chem.B 108,6616(2004).
[46]K.Andersson,P.?A.Malmqvist,B.O.Roos,A.J. Sadlej,and K.Wolinski,J.Phys.Chem.94,5483 (1990).
[47]K.Andersson,P.?A.Malmqvist,and B.O.Roos,J. Chem.Phys.96,1218(1992).
[48]N.′Forsberg and P.Malmqvist,Chem.Phys.Lett.274, 196(1997).
[49]F.Aquilante,R.Lindh,and T.B.Pedersen,J.Chem. Phys.127,114107(2007).
[50]G.Ghigo,B.O.Roos,and P.?A.Malmqvist,Chem. Phys.Lett.396,142(2004).
[51]W.H.Fang,J.Am.Chem.Soc.121,8376(1999).
[52]H.Y.He and W.H.Fang,J.Am.Chem.Soc.125, 16139(2003).
[53]W.H.Fang,Acc.Chem.Res.41,452(2008).
[54]G.L.Cui,L.Ding,F.Feng,Y.J.Liu,and W.H.Fang, J.Chem.Phys.132,194308(2010).
[55]G.L.Cui and W.H.Fang,ChemPhysChem 12,1689 (2011).
[56]G.L.Cui and W.H.Fang,ChemPhysChem 12,1351 (2011).
[57]G.L.Cui,Z.G.Sun,and W.H.Fang,J.Phys.Chem. A 115,10146(2011).
[58]G.L.Cui and W.H.Fang,J.Chem.Phys.138,044315 (2013).
[59]G.L.Cui and W.Thiel,J.Phys.Chem.Lett.5,2682 (2014).
[60]T.Yanai,D.Tew,and N.Handy,Chem.Phys.Lett. 393,51(2004).
[61]S.Vosko,L.Wilk,and M.Nusair,Can.J.Phys.58, 1200(1980).
[62]A.D.Becke,Phys.Rev.A 38,3098(1988).
[63]C.Lee,W.Yang,and R.Parr,Phys.Rev.B 37,785 (1988).
[64]A.D.Becke,J.Chem.Phys.98,1372(1993).
[65]R.Ditch fi eld,W.Hehre,and J.Pople,J.Chem.Phys. 54,724(1971).
[66]P.Hariharan and J.Pople,Theor.Chem.Acc.28,213 (1973).
[67]M.J.Frisch,G.W.Trucks,H.B.Schlegel,G.E. Scuseria,M.A.Robb,J.R.Cheesem,G.Scalmani, V.Barone,B.Mennucci,G.A.Petersson,H.Nakatsuji,M.Caricato,X.Li,H.P.Hratchian,A.F.Izmaylov,J.Bloino,G.Zheng,J.L.Sonnenberg,M. Hada,M.Ehara,K.Toyota,R.Fukuda,J.Hasegawa, M.Ishida,T.Nakajima,Y.Honda,O.Kitao,H.Nakai, T.Vreven,J.A.Montgomery Jr.,J.E.Peralta,F. Ogliaro,M.Bearpark,J.J.Heyd,E.Brothers,K.N. Kudin,V.N.Staroverov,R.Kobayashi,J.Normand, K.Raghavachari,A.Rendell,J.C.Burant,S.S.Iyengar,J.Tomasi,M.Cossi,N.Rega,J.M.Millam,M. Klene,J.E.Knox,J.B.Cross,V.Bakken,C.Adamo,J. Jaramillo,R.Gomperts,R.E.Stratmann,O.Yazyev, A.J.Austin,R.Cammi,C.Pomelli,J.W.Ochterski,R.L.Martin,K.Morokuma,V.G.Zakrzewski,G. A.Voth,P.Salvador,J.J.Dannenberg,S.Dapprich, A.D.Daniels,¨O.Farkas,J.B.Foresman,J.V.Ortiz, J.Cioslowski,D.J.Fox,Gaussian 09,Revision B.01. Wallingford CT:Gaussian,Inc.,(2010).
[68]F.Aquilante,L.De Vico,N.Ferr′e,G.Ghigo,P. Malmqvist,P.Neogr′ady,T.Pedersen,M.Pito?n′ak,M. Reiher,B.Roos,L.Serrano-Andr`es,M.Urban,V. Veryazov,and R.Lindh,J.Comput.Chem.31,224 (2010).
[69]J.Kouteck′y,V.Bona?ci′c-Kouteck′y,J.?C′??zek,D.D¨o, Int.J.Quantum Chem.12,357(1978).
[70]L.Salem,Acc.Chem.Res.12,87(1979).
[71]A.Viel,R.P.Krawczyk,U.Manthe,and W.Domcke, Angew.Chem.Int.Ed.42,3434(2003).
[72]G.L.Cui,P.J.Guan,and W.H.Fang,J.Phys.Chem. A 118,4732(2014).
[73]G.Cui,Z.Lan,and W.Thiel,J.Am.Chem.Soc.134, 1662(2012).
[74]L.Sp¨orkel,G.L.Cui,and W.Thiel,J.Phys.Chem.A 118,4732(2014).
 CHINESE JOURNAL OF CHEMICAL PHYSICS2016年1期
CHINESE JOURNAL OF CHEMICAL PHYSICS2016年1期
- CHINESE JOURNAL OF CHEMICAL PHYSICS的其它文章
- ARTICLE E ffi cient Separation of Ar and Kr from Environmental Samples for Trace Radioactive Noble Gas Detection?
- REVIEW Polarization Dependent Time-Resolved Infrared Spectroscopy and Its Applications?
- ARTICLE Reactions of Group V Metal Atoms with Hydrogen Sul fi de:Argon Matrix Infrared Spectra and Theoretical Calculations?
- ARTICLE Structural Dynamics of Phenyl Azide in Light-Absorbing Excited States: Resonance Raman and Quantum Mechanical Calculation Study?
- ARTICLE Structural and Infrared Spectroscopic Study on Solvation of Acetylene by Protonated Water Molecules?
- ARTICLE Infrared Photodisssociation Spectroscopy of Boron Carbonyl Cation Complexes?