
Properties and feasibility of using cancer stem cells in clinical cancer treatment
2017-01-13 01:54:49XiaoMeiGaoRuiZhangQiongZhuDongLunXiuQin
Cancer Biology & Medicine 2016年4期
Xiao-Mei Gao, Rui Zhang, Qiong-Zhu Dong,, Lun-Xiu Qin,
1Institutes of Biomedical Sciences, Fudan University, Shanghai 200032, China;2Department of General Surgery, Huashan Hospital and Cancer Metastasis Institute, Fudan University, Shanghai 200040, China
Properties and feasibility of using cancer stem cells in clinical cancer treatment
Xiao-Mei Gao1, Rui Zhang2, Qiong-Zhu Dong1,2, Lun-Xiu Qin1,2
1Institutes of Biomedical Sciences, Fudan University, Shanghai 200032, China;2Department of General Surgery, Huashan Hospital and Cancer Metastasis Institute, Fudan University, Shanghai 200040, China
Cancer treatment failure, drug resistance, or metastatic recurrence are thought to be caused mainly by the existence of a very small number of cancer stem cells (CSCs). The characteristics of this subgroup of cells include self-renewal, tumorigenesis, multiple differentiation and high invasiveness, metastasis, and drug resistance potential. Many studies have demonstrated that CSCs play important roles in tumor growth, spread and metastatic relapse after treatment, and are closely related to the prognosis of patients. From a therapeutic viewpoint, deep insights into the CSCs biology, development of specific therapeutic strategies for targeting CSCs, and characterization of their microenvironment could be an ideal way to combat cancer.
Cancer stem cells; cancers; treatment failure; metastasis; cancer therapy
Introduction
As one of the most serious diseases that afflict humans, cancer accounts for 15% of global mortality each year. Furthermore, 90% of patients with solid tumor die of metastasis, which consists of multiple processes associated with many factors, including epithelial-mesenchymal transition (EMT) and cancer stem cells (CSCs)1. Even in a hematopoietic system, where the cancers can be eradicated, cancer relapse always occurs2. Although patients are diagnosed as disease-free after cancer treatment, cancer frequently recurs several months or years later because of the redifferentiation and colonization of a small group of CSCs. Thus, at present, CSCs are considered as the cause of tumor initiation, relapse, metastasis, and drug resistance.
The exploration of CSCs can be traced back to the experiment on teratomas in the 18th century. At present, CSCs are widely considered as the root of tumors. Hematopoietic malignancies are the focus of basic research and clinical trials on CSCs. However, over the past several decades, with respect to stem cell biology, an effective and satisfactory cure for cancer has not been developed yet. From this perspective, the complexity of CSCs is considered as the main barrier for curing cancer. Thus, the origins, functions during tumor metastasis, and differentiation of CSCs should be revealed with urgency. Cutting-edge studies on CSCs have already progressed from seeking and screening related biomarkers to placing greater emphasis on the mechanism of molecular regulation and clinical transformation.
Until today, some CSCs biological features, which are regarded as distinct from the group’s parental counterparts, continue to be revealed. CSCs always remain quiescent to suppress their own outgrowth and to maintain their longterm survival until the stress from extra- or intra-tumor cells interferes and induces them to enter the cell cycle from G0/G1phase to aberrant rapid proliferation3,4. Moreover, some signal pathways, such as phosphatidylinositol 3-kinase (PI3K)/AKT5, interleukin 6 (IL6)/stat36, and Wnt/β-catenin7, which are closely associated with drug resistance, are abnormally activated. After CSCs or non-CSCs transform via an EMT and enter into circulation to become circulating tumor cells (CTCs), many proteins are ectopically expressed for the purposes of anti-apoptosis, immune escape, and even dormancy8,9. Currently, many clinical trials and pre-clinical experiments that target CSCs are ongoing. Molecular and pharmacological targeting of proline-rich tyrosine kinase 2 specifically induces cell death in aldehyde dehydrogenase (ALDH)-positive myeloma CSCs10. However, CSCs could not be completely destroyed, and they allow the regrowth of tumor because of uncovered complicated characteristics.Therefore, CSCs are the cause of cancer treatment failure; on the other hand, CSCs could be promising clinical targets for improving future cancer therapy11.
In this review, we discuss the role of CSCs in seeding tumor and the possible strategies for using them in clinical cancer treatment (Figure 1).
Overview of CSCs
The theory on CSCs was proposed in 1967 by Bergsagel12, who discovered that leukemic stem cells might exist in Philadelphia chromosome positive chronic myelogenous leukemia (CML). This observation challenged the traditional Darwinian “clonal selection theory, ” which advocates that tumor cells are derived from a single cell in the subclone13. During the past decades, more researchers have mentioned that when a group of sub-clonal cells are on the verge of extermination, about 5% of these cells narrowly escape apoptosis and acquire more malignant phenotypic properties such as tumorigenesis, metastasis, self-renewal, and drug resistance. Eventually, in 1997, Bonnet et al.14ascertained for the first time the existence of CSCs with the marker for CD34+/CD38-in acute myeloid leukemia (AML). In 2003, Al-Hajj et al.15successfully obtained CSCs from breast carcinoma by using the marker for CD44+/CD24-. Thereafter, CSCs were found in other kinds of solid malignancies, including those of the brain16and the liver17.
Although increasing evidence has indicated the existence of CSCs, the origin of CSCs remains unknown. At present, three hypotheses are used to explain the origin of CSCs18,19. First, adult stem cells or progenitor cells from normal tissues become CSCs because of somatic mutation accumulation at the genetic or epigenetic level20,21, or differentiated tumor cells become CSCs through EMT. This hypothesis attracts more researchers to exert effort on discovering the differences between CSCs and transformed cells. Learning about such differences may help modify current cancer treatments more precisely or even develop a completely effective cure for cancer. Second, the differentiated cell and tissue stem cell fuse into a euploidy cell. The euploidy cell has an unstable genome, which is prone to mutating, and it can finally transform into CSCs. This phenomenon is a rare event, but it might contribute to tumor progression. Third, the normal cells absorb exogenous DNA from nearby tumor cells and integrate the genetic material into their own genomes, resulting in the suppression or ectopic expression of some genes and ultimately leading to the transformation of CSCs. This process is not easily recognized. Given the diversity of the hypotheses, the origin of CSCs still remains a fundamental issue to be addressed.
CSCs: controversial biomarkers in solid tumor
The CSCs hypothesis contradicts the classical theory of clonal selection in which only CSCs is considered as the origin and seed of tumors. However, the heterogeneity of CSCs is universal. Thus, selecting a single specific CSCs biomarker for a certain type of cancer is arduous. For example, CD138-22, ALDH1+23, and side population cells have been used to identify multiple myeloma stem cells. CD13324,25, CD2426, CD9027, and CD4728are all approved to be the markers in hepatic CSCs; leucine-rich repeat-containing G-protein coupled receptor 5 (Lgr5)29, CD13330, and CD4431are used in colonic carcinoma. These markers indicate that distingui-shing CSCs from other kinds of cancer cells by surface biomarkers alone is difficult. This confusion may result from the limitation of the current detection techniques and understanding of CSCs.
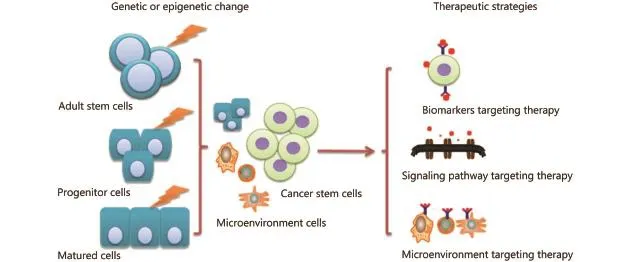
Figure 1 Origin, regulating mechanisms, and possible targeted therapy for CSCs.
The transformation of CSCs can be more suitably described as the clonal evolution of CSCs to survive in their changing microenvironment. The different biomarkers indicate that different potentials of CSCs emerge under certain conditions, including the accumulation of mutation, which maintains the long-term survival of CSCs. Thus, the CSCs model and clonal evolution model in cancers could coexist. In fact, in some cancers, CSCs hypothesis is not considered because among cancer cells, the intrinsic differences, which are characterized as genetic and epigenetic differences among tumorigenic cancer cells that lack hierarchical organization32, can be explained by clonal evolution. In CML patients who relapsed after sequential treatment with the ABL inhibitors, imatinib and dasatinib, resistant BCR-ABL kinase domain mutations, including T315I, are revealed in the CSCs33.
CSCs: seeds of metastasis
Given the strong potential of the group, CSCs are regarded as the seeds of metastasis. When CSCs disseminate into circulation, they may become a part of CTCs. Recently, many studies suggest that the portion of CTCs that can successfully form metastases may as well be CSCs or have some characteristics of CSCs. In breast cancer, a part of epithelial cell adhesion molecule (EpCAM)+CTCs expresses CD133+or ATP-binding cassette sub-family G member 2+, hinting that CSCs could be the major functional cells in all CTCs, or only the CTCs with the characteristics of CSCs could survive metastasis34. Moreover, the patients who have CTCs with the phenotype or molecular type of CSCs have a more dismal prognosis34. When metastasis progresses, CSCs in circulation prepare to enter self-driven quiescence and decrease the expression of ligands recognized by the immune system to survive. In breast cancer, metastatic stem cells highly express sex-determining region Y-box (SOX)2 or SOX9 and upregulate dickkopf-related protein 1, inhibiting the Wnt signaling pathway and downregulating the natural killer cell ligands35. In colorectal cancer, CD110+intestinal CSCs have a strong potential for liver metastasis36. The thrombopoietin produced by the liver is a ligand of CD110, which induces Wnt pathway activation and promotes the self-renewal of CSCs9. In colon cancer, reports showed that the expression of doublecortin-like kinase 1 and Lgr5 are upregulated in both CSCs and CTCs37. Thus, many scientists combine CTCsrelated research with the use of CSCs.
The communication between CSCs and the microenvironment in primary tumor affects the stemness factors or pathways facilitating the invasiveness and metastatic abilities of CSCs38-41. After the formation of premetastases, some attractors could further guide CSCs to go“home” to specific distal tissues. Finally, the changing metabolism of CSCs and the cross-talk of the cells with the new environment during the formation of metastatic cells also regulate the self-renewal and settlement of CSCs. The researchers aim to find intrinsic driving factors within these processes and to perform more effective targeted therapies. In hepatocellular carcinoma, when CD44+CSCs are cocultured with CD14+tumor-associated macrophage (TAM), their migration and tumorigenic abilities are enhanced by IL6 from TAM42. In breast cancer, when CD90+cells interact with TAM, EphA4 is upregulated to modulate the activity of CD90+breast carcinoma cells41. In addition, the exosome, which is secreted by mesenchymal cells in the primary tumor of pancreatic cancer, reaches the targeted organ in advance to remold the microenvironment, thereby facilitating the adaptability of metastatic pancreatic cancer cells and elevating their survival rate43.
Treatment strategies that target CSCs
After cytotoxic treatments, such as chemotherapy, radiotherapy, or targeted therapy, the tumor initially shrinks or disappears; however, relapse may occur and cause the death of cancer patients44. The theory on CSCs tries to identify the reason for this phenomenon. CSCs are considered as the “beating heart” of tumors. Thus, possibly, only by exterminating CSCs could cancer be cured. Actually CSCs can be targeted through various strategies, such as by inhibiting the molecular pathway(s) of CSCs45,46, targeting CSCs or the microenvironment47, combining CSC-targeted therapy with chemotherapy, or differentiating inducers to promote the differentiation of CSCs into bulk tumor cells, which can be eradicated by chemotherapy48(Table 1).
Targeted treatments always focus on the cell surface or abnormally activated signaling pathway of CSCs or the cells in the niche. CD13349, CD1350, and CD2451could be targets for eliminating CSCs. The embryonic signaling pathways always abnormally activate CSCs, which are inhibited by different inhibitors, such as cyclopamine, the prototype of Hedgehog (Hh) pathway-specific inhibitors52; iCG001, which selectively inhibits Wnt/β-catenin signaling53; and γ-secretase inhibitors, which block notch signaling54. Adoptive therapy with ALDH1 family member A1 (ALDH1A1)-specific CD8+ T cells eliminates ALDH bright cells, which have thecharacteristics of cancer-initiating cells. The curative effect of classical treatments may be increased when combined with the targeted therapy of CSCs. CSCs resist drugs because of their quiescent state, whereas most drug treatments target proliferating cells. This characteristic suggests that stimulating CSCs to differentiate may increase their sensitivity to drugs. Oncostatin M induces EpCAM+liver CSCs to differentiate into a hepatocytic lineage, causing the CSCs to be more sensitive to 5-fluorouracil55.
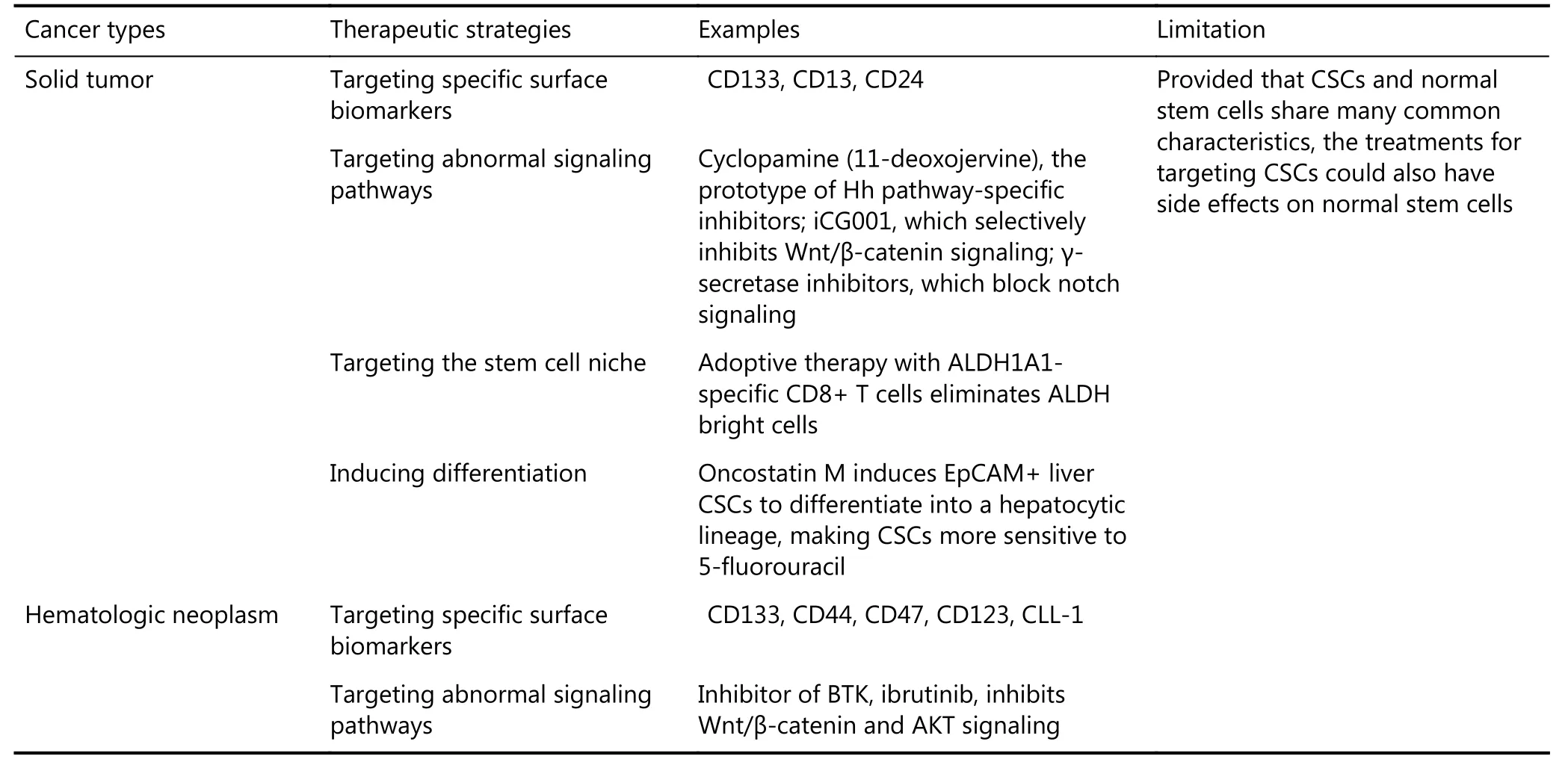
Table 1 Treatment strategies for targeting CSCs
In a hematological system, given that CSCs were first identified in AML in 1994, eradicating the leukemia stem cells (LSCs) has been regarded as the optimal way of decreasing the possibility of relapse. Similar to the solid tumor, LSCs can be targeted with two kinds of treatment strategies, such as by interfering in the abnormal signal pathways or by targeting the specific surface biomarkers of LCSs56,57. Along with their unique characteristics, distinct alterations in many signal pathways, such as Wnt, Notch, NF-κB, and PI3K-AKT, which play important roles in cell self-renewal, proliferation, motility, stemness maintenance, and survival, have been identified in LSCs compared with their normal corresponding hematopoietic stem cells (HSCs). Some inhibitors, such as asbortezomib, parthenolide, and rapamycin, which target abnormally activated pathways, have been developed. Bruton tyrosine kinase (BTK), which could govern or at least participate in Wnt/β-catenin and AKT signaling in myeloma stemness, is not expressed in normal plasma cells. Thus, the BTK inhibitors, including ibrutinib or CGI1746, are promising therapeutic candidates58. However, the major concern regarding these inhibitors is their potential toxicity to the normal hematopoietic system because these pathways are also functional in HSCs. Thus, discovering a target that could selectively impair the survival, self-renewal, or proliferation pathways of LSCs is important.
The discovery of the specific surface molecules of LSCs provides some important insights. An ideal therapeutic target should be expressed on LSCs but not on the normal HSCs. Several candidate antigens, such as CD33, CD44, CD47, CD123, and CLL-1, have been identified59,60. The monoclonal antibodies against these markers show more specificity in in vitro experiments than the inhibitors of signal pathways61. However, the expression levels of certain LSCs surface markers may vary greatly in patients. Even within the same patient, more than one phenotypically distinct subpopulation of LSCs might be found. This information suggests that the cell surface antigen profiles of LSCs may vary. Another challenge in the treatment of hematological malignancies is that targeted drugs cannot reach the appropriate concentration in circulation to induce an effective action. To deal with this problem, the surface markers are combined with nano-delivery systems. The multifunctional delivery using targeting ligands (e.g., hyaluronic acid to target CD44) and other combination therapeutics (e.g., siRNA with small molecular drug) can enhance the treatment efficacy62.
CSCs and normal stem cells share many common characteristics, including stemness-related gene expression profiles and capabilities for self-renewal, development into lineages, and proliferation. Thus, the inhibitors that target CSCs could also have side effects on normal stem cells63. The side effects may be due to the inaccurate experimental methods that simply regard CSCs as adult stem cells. However, non-CSCs progeny could revert to CSCs under certain conditions, implying the dynamic transition between CSCs and non-CSCs64. In addition, multi-lineage differentiation is not a fixed feature of CSCs, because most papers published in recent years have shown that CSCs may only possess mono-potential, which could result in non-CSCs. However, if all of these key points are addressed, specific targeting of CSCs is possible. Robert A. Weinberg, an expert in CSCs-targeted therapy, is taking the risk with treatment through his Verastem Incorporated, which has over 2 trillion dollars in investment and is conducting more than 60 CSCs-related clinical trials65.
Problems and prospects
CSCs models have been established for most types of cancer, and related discoveries have revealed important information on them. However, their applications in clinical practices still face many obstacles. First, given the uncertainty of CSCsspecific biomarkers, the criteria for CSCs are not simple and accurate enough. Second, the gold standard for approving CSCs is introducing them to immuno-deficient mice at various doses by injection, which is time-consuming and expensive. Thus, administration is another difficulty in the study of CSCs. Third, the origin of CSCs may vary, and a dynamic transition exists between CSCs and non-CSCs. Therefore, eradicating the existing CSCs does not guarantee that non-CSCs will never transform into CSCs. Furthermore, provided that human cancer is a systemic disease under unbalanced homeostasis, the passive attitudes demonstrate that tumors may not disappear even after CSCs are eliminated. In consequence, although CSCs are promising therapeutic targets, more comprehensive studies are needed to uncover all myths involved in CSCs and to establish a bridge between the progress in basic studies and clinical application in routine cancer diagnosis and treatment of CSCs.
Acknowledgments
This work is supported by the grants from the National Key Basic Research Program of China (Grant No. 2013CB910500), China National Key Projects for Infectious Disease (Grant No. 2012ZX10002-012), and National Natural Science Foundation of China (Grant No. 81372647).
Conflict of interest statement
No potential conflicts of interest are disclosed
1.Gupta GP, Massagué J. Cancer metastasis: building a framework. Cell. 2006; 127: 679-95.
2.Delude C. Tumorigenesis: testing ground for cancer stem cells. Nature. 2011; 480: S43-5.
3.Chen X, Li X, Zhao BH, Shang DH, Zhong M, Deng CF, et al. Dormancy activation mechanism of oral cavity cancer stem cells. Tumour Biol. 2015; 36: 5551-9.
4.Li L, Bhatia R. Stem cell quiescence. Clin Cancer Res. 2011; 17: 4936-41.
5.Xu YQ, Jiang ZM, Zhang ZH, Sun NN, Zhang M, Xie J, et al. HtrA1 downregulation induces cisplatin resistance in lung adenocarcinoma by promoting cancer stem cell-like properties. J Cell Biochem. 2014; 115: 1112-21.
6.Zhong HH, Davis A, Ouzounova M, Carrasco RA, Chen C, Breen S, et al. A novel IL6 antibody sensitizes multiple tumor types to chemotherapy including trastuzumab-resistant tumors. Cancer Res. 2016; 76: 480-90.
7.Fong CY, Gilan O, Lam EYN, Rubin AF, Ftouni S, Tyler D, et al. BET inhibitor resistance emerges from leukaemia stem cells. Nature. 2015; 525: 538-42.
8.Sosa MS, Parikh F, Maia AG, Estrada Y, Bosch A, Bragado P, et al. NR2F1 controls tumour cell dormancy via SOX9- and RARβdriven quiescence programmes. Nat Commun. 2015; 6: 6170.
9.Wu ZM, Wei D, Gao WC, Xu YT, Hu ZQ, Ma ZY, et al. TPO-induced metabolic reprogramming drives liver metastasis of colorectal cancer CD110+tumor-initiating cells. Cell Stem Cell. 2015; 17: 47-59.
10.Meads MB, Fang B, Mathews L, Gemmer J, Nong L, Rosado-Lopez I, et al. Targeting PYK2 mediates microenvironment-specific cell death in multiple myeloma. Oncogene. 2016; 35: 2723-34.
11.Zhao JJ, Lin JH, Zhu D, Wang XJ, Brooks D, Chen M, et al. miR-30-5p functions as a tumor suppressor and novel therapeutic tool by targeting the oncogenic Wnt/β-catenin/BCL9 pathway. Cancer Res. 2014; 74: 1801-13.
12.Bergsagel DE. The chronic leukemias: a review of disease manifestations and the aims of therapy. Can Med Assoc J. 1967; 96: 1615-20.
13.Shackleton M, Quintana E, Fearon ER, Morrison SJ. Heterogeneity in cancer: cancer stem cells versus clonal evolution. Cell. 2009; 138: 822-9.
14.Bonnet D, Dick JE. Human acute myeloid leukemia is organized asa hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997; 3: 730-7.
15.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003; 100: 3983-8.
16.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. Identification of human brain tumour initiating cells. Nature. 2004; 432: 396-401.
17.Yamashita T, Ji JF, Budhu A, Forgues M, Yang W, Wang HY, et al. EpCAM-positive hepatocellular carcinoma cells are tumorinitiating cells with stem/progenitor cell features. Gastroenterology. 2009; 136: 1012-24.
18.Blanpain C. Tracing the cellular origin of cancer. Nat Cell Biol. 2013; 15: 126-34.
19.Bjerkvig R, Tysnes BB, Aboody KS, Najbauer J, Terzis AJA. Opinion: the origin of the cancer stem cell: current controversies and new insights. Nat Rev Cancer. 2005; 5: 899-904.
20.Sutherland KD, Proost N, Brouns I, Adriaensen D, Song JY, Berns A. Cell of origin of small cell lung cancer: inactivation of Trp53 and Rb1 in distinct cell types of adult mouse lung. Cancer Cell. 2011; 19: 754-64.
21.Chakrabarti R, Wei Y, Hwang J, Hang X, Andres Blanco M, Choudhury A, et al. ΔNp63 promotes stem cell activity in mammary gland development and basal-like breast cancer by enhancing Fzd7 expression and Wnt signalling. Nat Cell Biol. 2014; 16: 1004-15, 1-13.
22.Matsui W, Huff CA, Wang QJ, Malehorn MT, Barber J, Tanhehco Y, et al. Characterization of clonogenic multiple myeloma cells. Blood. 2004; 103: 2332-6.
23.Zhou W, Yang Y, Gu Z, Wang H, Xia J, Wu X, et al. ALDH1 activity identifies tumor-initiating cells and links to chromosomal instability signatures in multiple myeloma. Leukemia. 2014; 28: 1155-8.
24.Hou Y, Zou QF, Ge RL, Shen F, Wang YZ. The critical role of CD133+CD44+/high tumor cells in hematogenous metastasis of liver cancers. Cell Res. 2012; 22: 259-72.
25.Tang KH, Ma S, Lee TK, Chan YP, Kwan PS, Tong CM, et al. CD133+liver tumor-initiating cells promote tumor angiogenesis, growth, and self-renewal through neurotensin/interleukin-8/CXCL1 signaling. Hepatology. 2012; 55: 807-20.
26.Lee TKW, Castilho A, Cheung VCH, Tang KH, Ma S, Ng IOL. CD24+ liver tumor-initiating cells drive self-renewal and tumor initiation through STAT3-mediated NANOG regulation. Cell Stem Cell. 2011; 9: 50-63.
27.Yang ZF, Ho DW, Ng MN, Lau CK, Yu WC, Ngai P, et al. Significance of CD90+cancer stem cells in human liver cancer. Cancer Cell. 2008; 13: 153-66.
28.Lee TKW, Cheung VCH, Lu P, Lau EYT, Ma S, Tang KH, et al. Blockade of CD47-mediated cathepsin S/protease-activated receptor 2 signaling provides a therapeutic target for hepatocellular carcinoma. Hepatology. 2014; 60: 179-91.
29.Schepers AG, Snippert HJ, Stange DE, van den Born M, van Es JH, van de Wetering M, et al. Lineage tracing reveals Lgr5+ stem cell activity in mouse intestinal adenomas. Science. 2012; 337: 730-5.
30.Vincent Z, Urakami K, Maruyama K, Yamaguchi K, Kusuhara M. CD133-positive cancer stem cells from Colo205 human colon adenocarcinoma cell line show resistance to chemotherapy and display a specific metabolomic profile. Genes Cancer. 2014; 5: 250-60.
31.Ohata H, Ishiguro T, Aihara Y, Sato A, Sakai H, Sekine S, et al. Induction of the stem-like cell regulator CD44 by Rho kinase inhibition contributes to the maintenance of colon cancerinitiating cells. Cancer Res. 2012; 72: 5101-10.
32.Klevebring D, Rosin G, Ma R, Lindberg J, Czene K, Kere J, et al. Sequencing of breast cancer stem cell populations indicates a dynamic conversion between differentiation states in vivo. Breast Cancer Res. 2014; 16: R72.
33.Shah NP, Skaggs BJ, Branford S, Hughes TP, Nicoll JM, Paquette RL, et al. Sequential ABL kinase inhibitor therapy selects for compound drug-resistant BCR-ABL mutations with altered oncogenic potency. J Clin Invest. 2007; 117: 2562-9.
34.Sun YF, Xu Y, Yang XR, Guo W, Zhang X, Qiu SJ, et al. Circulating stem cell-like epithelial cell adhesion molecule-positive tumor cells indicate poor prognosis of hepatocellular carcinoma after curative resection. Hepatology. 2013; 57: 1458-68.
35.Malladi S, Macalinao DG, Jin X, He L, Basnet H, Zou YL, et al. Metastatic latency and immune evasion through autocrine inhibition of WNT. Cell. 2016; 165: 45-60.
36.Gao WC, Chen L, Ma ZY, Du ZG, Zhao ZH, Hu ZQ, et al. Isolation and phenotypic characterization of colorectal cancer stem cells with organ-specific metastatic potential. Gastroenterology. 2013; 145: 636-46.
37.Mirzaei A, Tavoosidana G, Rad AA, Rezaei F, Tavakoli-Yaraki M, Kadijani AA, et al. A new insight into cancer stem cell markers: could local and circulating cancer stem cell markers correlate in colorectal cancer? Tumour Biol. 2016; 37: 2405-14.
38.Brooks MD, Wicha MS. Tumor twitter: cellular communication in the breast cancer stem cell niche. Cancer Discov. 2015; 5: 469-71.
39.Zhang M, Tsimelzon A, Chang CH, Fan C, Wolff A, Perou CM, et al. Intratumoral heterogeneity in a Trp53-null mouse model of human breast cancer. Cancer Discov. 2015; 5: 520-33.
40.Cojoc M, M?bert K, Muders MH, Dubrovska A. A role for cancer stem cells in therapy resistance: cellular and molecular mechanisms. Semin Cancer Biol. 2015; 31: 16-27.
41.Lu HH, Clauser KR, Tam WL, Fr?se J, Ye X, Eaton EN, et al. A breast cancer stem cell niche supported by juxtacrine signalling from monocytes and macrophages. Nat Cell Biol. 2014; 16: 1105-17.
42.Wan SS, Zhao ED, Kryczek I, Vatan L, Sadovskaya A, Ludema G, et al. Tumor-associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology. 2014; 147: 1393-404.
43.Costa-Silva B, Aiello NM, Ocean AJ, Singh S, Zhang HY, Thakur BK, et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat Cell Biol. 2015; 17: 816-26.
44.Bao SD, Wu QL, McLendon RE, Hao YL, Shi Q, Hjelmeland AB, etal. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006; 444: 756-60.
45.Takebe N, Harris PJ, Warren RQ, Ivy SP. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat Rev Clin Oncol. 2011; 8: 97-106.
46.Abetov D, Mustapova Z, Saliev T, Bulanin D, Batyrbekov K, Gilman CP. Novel small molecule inhibitors of cancer stem cell signaling pathways. Stem Cell Rev. 2015; 11: 909-18.
47.Visus C, Wang YY, Lozano-Leon A, Ferris RL, Silver S, Szczepanski MJ, et al. Targeting ALDHbright human carcinoma-initiating cells with ALDH1A1-specific CD8+ T cells. Clin Cancer Res. 2011; 17: 6174-84.
48.Merino VF, Nguyen N, Jin K, Sadik H, Cho S, Korangath P, et al. Combined treatment with epigenetic, differentiating, and chemotherapeutic agents cooperatively targets tumor-initiating cells in triple-negative breast cancer. Cancer Res. 2016; 76: 2013-24.
49.Lan X, Wu YZ, Wang Y, Wu FR, Zang CB, Tang C, et al. CD133 silencing inhibits stemness properties and enhances chemoradiosensitivity in CD133-positive liver cancer stem cells. Int J Mol Med. 2013; 31: 315-24.
50.Christ B, Stock P, Dollinger MM. CD13: waving the flag for a novel cancer stem cell target. Hepatology. 2011; 53: 1388-90.
51.He H, Tu XJ, Zhang J, Acheampong DO, Ding L, Ma ZX, et al. A novel antibody targeting CD24 and hepatocellular carcinoma in vivo by near-infrared fluorescence imaging. Immunobiology. 2015; 220: 1328-36.
52.Taipale J, Chen JK, Cooper MK, Wang BL, Mann RK, Milenkovic L, et al. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature. 2000; 406: 1005-9.
53.Emami KH, Nguyen C, Ma H, Kim DH, Jeong KW, Eguchi M, et al. A small molecule inhibitor of β-catenin/cyclic AMP response element-binding protein transcription. Proc Natl Acad Sci USA. 2004; 101: 12682-7.
54.Rizzo P, Osipo C, Foreman K, Golde T, Osborne B, Miele L. Rational targeting of Notch signaling in cancer. Oncogene. 2008; 27: 5124-31.
55.Yamashita T, Honda M, Nio K, Nakamoto Y, Yamashita T, Takamura H, et al. Oncostatin m renders epithelial cell adhesion molecule-positive liver cancer stem cells sensitive to 5-Fluorouracil by inducing hepatocytic differentiation. Cancer Res. 2010; 70: 4687-97.
56.Crews LA, Jamieson CHM. Selective elimination of leukemia stem cells: hitting a moving target. Cancer Lett. 2013; 338: 15-22.
57.Krause DS, Van Etten RA. Right on target: eradicating leukemic stem cells. Trends Mol Med. 2007; 13: 470-81.
58.Yang Y, Shi JM, Gu ZM, Salama ME, Das S, Wendlandt E, et al. Bruton tyrosine kinase is a therapeutic target in stem-like cells from multiple myeloma. Cancer Res. 2015; 75: 594-604.
59.Jordan CT. Targeting myeloid leukemia stem cells. Sci Transl Med. 2010; 2: 31ps21.
60.Majeti R. Monoclonal antibody therapy directed against human acute myeloid leukemia stem cells. Oncogene. 2011; 30: 1009-19.
61.Taussig DC, Miraki-Moud F, Anjos-Afonso F, Pearce DJ, Allen K, Ridler C, et al. Anti-CD38 antibody-mediated clearance of human repopulating cells masks the heterogeneity of leukemia-initiating cells. Blood. 2008; 112: 568-75.
62.Iyer AK, Singh A, Ganta S, Amiji MM. Role of integrated cancer nanomedicine in overcoming drug resistance. Adv Drug Deliv Rev. 2013; 65: 1784-802.
63.Pardal R, Clarke MF, Morrison SJ. Applying the principles of stemcell biology to cancer. Nat Rev Cancer. 2003; 3: 895-902.
64.Santisteban M, Reiman JM, Asiedu MK, Behrens MD, Nassar A, Kalli KR, et al. Immune-induced epithelial to mesenchymal transition in vivo generates breast cancer stem cells. Cancer Res. 2009; 69: 2887-95.
65.Kaiser J. The cancer stem cell gamble. Science. 2015; 347: 226-9.
Cite this article as:Gao X, Zhang R, Dong Q, Qin L. Properties and feasibility of using cancer stem cells in clinical cancer treatment. Cancer Biol Med. 2016; 13: 489-95. doi: 10.20892/j.issn.2095-3941.2016.0076
Lun-Xiu Qin
E-mail: qinlx@fudan.edu.cn
Received September 6, 2016; accepted October 24, 2016.
Available at www.cancerbiomed.org
Copyright ? 2016 by Cancer Biology & Medicine
 Cancer Biology & Medicine2016年4期
Cancer Biology & Medicine2016年4期
- Cancer Biology & Medicine的其它文章
- Work-up and management of a high-risk patient with primary central nervous system lymphoma
- Leptin influences estrogen metabolism and increases DNA adduct formation in breast cancer cells
- Ki-67 as a prognostic marker according to breast cancer molecular subtype
- Human endogenous retroviruses and cancer
- Molecular landscape in acute myeloid leukemia: where do we stand in 2016
- Surgical treatment of intrahepatic cholangiocarcinoma: a retrospective study of 104 cases