
Neuroprotective effects of ischemic preconditioning on hippocampal CA1 pyramidal neurons through maintaining calbindin D28k immunoreactivityfollowing subsequent transient cerebral ischemia
2017-08-07 02:26InHyeKimYongHwanJeonTaeKyeongLeeJeongHwiChoJaeChulLeeJoonHaParkJiHyeonAhnBichNaShinYangHeeKimSeongkweonHongBingChunYanMooHoWonYunLyulLee
中國神經(jīng)再生研究(英文版) 2017年6期
In Hye Kim, Yong Hwan Jeon, Tae-Kyeong Lee Jeong Hwi Cho Jae-Chul Lee Joon Ha Park, Ji Hyeon Ahn, Bich-Na Shin, Yang Hee Kim, Seongkweon Hong, Bing Chun Yan, Moo-Ho Won, Yun Lyul Lee,
1 Department of Neurobiology, School of Medicine, Kangwon National University, Chuncheon, South Korea
2 Department of Radiology, School of Medicine, Kangwon National University, Chuncheon, South Korea
3 Department of Biomedical Science, Research Institute of Bioscience and Biotechnology, Hallym University, Chuncheon, South Korea
4 Department of Physiology, College of Medicine, Hallym University, Chuncheon, South Korea
5 Department of Surgery, School of Medicine, Kangwon National University, Chuncheon, South Korea
6 Institute of Integrative Traditional & Western Medicine, Medical College, Yangzhou University, Yangzhou, Jiangsu Province, China
RESEARCH ARTICLE
Neuroprotective effects of ischemic preconditioning on hippocampal CA1 pyramidal neurons through maintaining calbindin D28k immunoreactivity
following subsequent transient cerebral ischemia
In Hye Kim1,#, Yong Hwan Jeon2,#, Tae-Kyeong Lee1, Jeong Hwi Cho1, Jae-Chul Lee1, Joon Ha Park3, Ji Hyeon Ahn3, Bich-Na Shin4, Yang Hee Kim5, Seongkweon Hong5, Bing Chun Yan6, Moo-Ho Won1,*, Yun Lyul Lee4,*
1 Department of Neurobiology, School of Medicine, Kangwon National University, Chuncheon, South Korea
2 Department of Radiology, School of Medicine, Kangwon National University, Chuncheon, South Korea
3 Department of Biomedical Science, Research Institute of Bioscience and Biotechnology, Hallym University, Chuncheon, South Korea
4 Department of Physiology, College of Medicine, Hallym University, Chuncheon, South Korea
5 Department of Surgery, School of Medicine, Kangwon National University, Chuncheon, South Korea
6 Institute of Integrative Traditional & Western Medicine, Medical College, Yangzhou University, Yangzhou, Jiangsu Province, China
Ischemic preconditioning elicited by a non-fatal brief occlusion of blood flow has been applied for an experimental therapeutic strategy against a subsequent fatal ischemic insult. In this study, we investigated the neuroprotective effects of ischemic preconditioning (2-minute transient cerebral ischemia) on calbindin D28k immunoreactivity in the gerbil hippocampal CA1 area following a subsequent fatal transient ischemic insult (5-minute transient cerebral ischemia). A large number of pyramidal neurons in the hippocampal CA1 area died 4 days aer 5-minute transient cerebral ischemia. Ischemic preconditioning reduced the death of pyramidal neurons in the hippocampal CA1 area. Calbindin D28k immunoreactivity was greatly attenuated at 2 days aer 5-minute transient cerebral ischemia and it was hardly detected at 5 days post-ischemia. Ischemic preconditioning maintained calbindin D28k immunoreactivity aer transient cerebral ischemia.ese findings suggest that ischemic preconditioning can attenuate transient cerebral ischemia-caused damage to the pyramidal neurons in the hippocampal CA1 area through maintaining calbindin D28k immunoreactivity.
nerve regeneration; transient cerebral ischemia; ischemic tolerance; neuroprotection; hippocampus; pyramidal neurons; calcium binding protein; neural regeneration
Introduction
Calcium ions play a complex and fatal role in nerve cell function (Cai et al., 2015). Evidence exists that uncontrolled increase in intracellular calcium ion concentrations results in excessive cell activation, injury, and eventually cell death, and increased calcium ion concentrations lead to calcium binding by regulatory proteins, which are called calcium binding proteins including calbindin, parvalbumin and calretinin (Baimbridge et al., 1992; Verdaguer et al., 2015).
Calbindin-D28k (CB), a member of the EF-hand family of calcium binding proteins, is easily found in neuronal cells in the central nervous system and performs function that is able to buffer toxic intracellular calcium ions induced by various insults including ischemic stroke (Baimbridge et al., 1992; Klapstein et al., 1998; Yenari et al., 2001). CB overexpression shows resistance to ischemic insultsin vitroandin vivo(Yenari et al., 2001; Fan et al., 2007; Freimann et al., 2010); and exogenous CB reduces oxidative stress, preserves mitochondrial function (Guo et al., 1998), and delays the onset of cell death following excitotoxic stimulation (D’Orlando et al., 2001).
Transient global cerebral ischemia, which is able to happen due to the blockage or lack of cerebral blood flow induced by cardiac arrest or cardiovascular surgery, leads to death of vulnerable neurons such as pyramidal neurons in the hippocampal CA1 area (Ohk et al., 2012; Lee et al., 2013a; Kim et al., 2014). Ischemic preconditioning (IPC), which is a technique for producing resistance to the loss of blood supply, can be elicited by non-fatal brief occlusion of blood flow, has been demonstrated as a therapeutic strategy against subsequent fatal ischemic insults (Lehotsky et al., 2009; Liu et al., 2009;ompson et al., 2013; Kovalska et al., 2014). IPC can activate certain cellular pathways that are able to help alleviate fatal damage induced by subsequent ischemic insults,and this phenomenon is called “ischemic tolerance” (Saad et al., 2015).
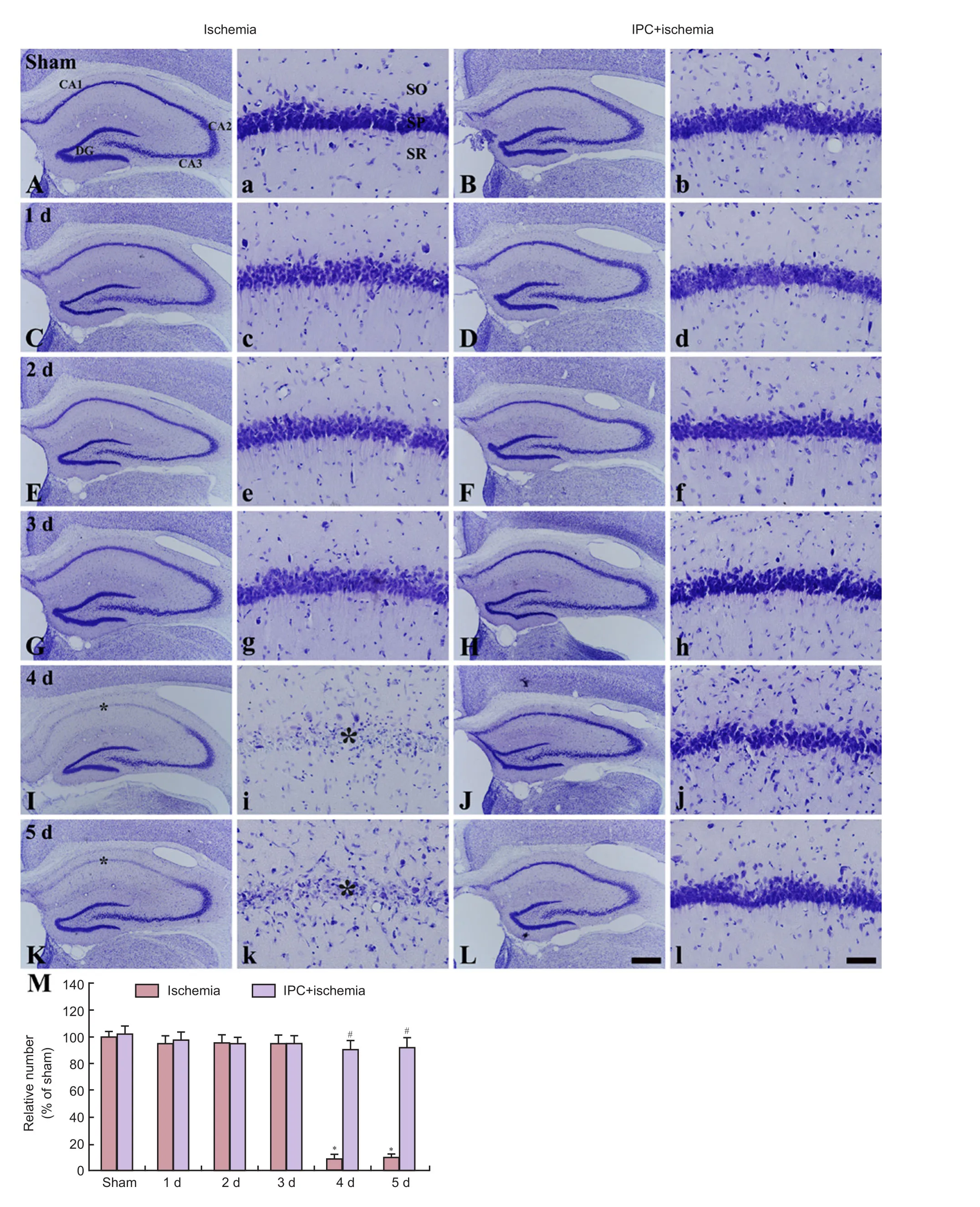
Figure 1 Cresyl violet (CV) staining of the gerbil hippocampus in the sham (A, a), IPC + sham (B, b), ischemia (C, c, E, e, G, g, I, i, K, and k), and IPC+ ischemia (D, d, F, f, H, h, J, j, L, and l) groups.
Several mechanisms, which explain the neuroprotective effects of IPC, have been published (Lee et al., 2014, 2015a, b, 2016; Kim et al., 2015; Park et al., 2016). However, to the best of our knowledge, there are no reports on calcium binding proteins in IPC-mediated ischemic brains. We, therefore, investigated the effects of IPC on CB immunoreactivity in the hippocampal CA1 pyramidal neurons following subsequent ischemia/reperfusion injury in gerbils.
Materials and Methods
Experimental animals
As previously described (Kim et al., 2015), male gerbils weighing 65–75 g and aged 6 months, were divided into four groups: (1) sham group (both common carotid arteries were exposed but not occluded); (2) ischemia group (5-minute transient ischemia); (3) IPC + sham group (IPC (2-minute transient ischemia) and no ischemia); and (4) IPC + ischemia group (IPC followed by ischemia). Gerbils (n= 7 at each point time in each group) were recovered 1, 2, 3, 4 and 5 days after ischemia. Procedures for animal handling and care complied with the guidelines that follow the current international laws and policies (Guide for the Care and Use of Laboratory Animals,e National Academies Press, 8th Ed., 2011), and all the experimental protocols of this study were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC), Kangwon National University (approval No. KW-160802-1).
Induction of IPC and transient ischemia
As previously described (Park et al., 2016), in brief, the gerbils were anesthetized with a mixture of 2.5% isoflurane (Baxtor, Deerfield, IL, USA) in 33% oxygen and 67% nitrous oxide. Bilateral common carotid arteries were occluded for 2 minutes for IPC followed by 5-minute ischemia with 1 day interval. Body (rectal) temperature was controlled under normothermia (37 ± 0.5°C) during the surgery with a rectal temperature probe (TR-100; Fine Science Tools, Foster City, CA, USA).
Tissue processing
Tissue preparation for histology was carried out according to our published method (Kim et al., 2014). In brief, the gerbils were perfused transcardially with 4% paraformaldehyde.e brains were serially sectioned into 30-μm coronal sections using a cryostat (Leica, Wetzlar, Germany).
Cresyl violet (CV) staining
In order to observe the distribution of pyramidal cells in the stratum pyramidale of hippocampal CA1 region, CV staining was done as previously described (Park et al., 2016). Briefly, 1% cresyl violet acetate (Sigma, St. Louis, MO, USA) and 0.28% glacial acetic acid were used for CV staining.
Fluoro-Jade B (F-J B) histofluorescence staining
To examine neuronal death after ischemia, F-J B histofluorescence staining was carried out according to our published procedure (Park et al., 2016). In short, the brain tissues were immersed in a 0.06 % potassium permanganate solution and stained with 0.0004% F-J B (Histochem, Jefferson, AR, USA) solution.e stained brain tissues were observed using an epifluorescent microscope (Carl Zeiss, Oberkochen, Germany) with blue (450–490 nm) excitation light and a barrier filter.
Immunohistochemistry for CB
CB immunoreactivity was determined according to our published method (Bae et al., 2015). Briefly, the brain tissues were incubated with diluted rabbit anti-CB antibody (1:500; Abcam, Cambridge, MA, USA) overnight at 4°C, then exposed to biotinylated goat anti-rabbit antibody (1:250; Vector Laboratories, Burlingame, CA, USA) for 2 hours at room temperature and streptavidin peroxidase complex (1:200; Vector Laboratories) and finally visualized with 3,3′-diaminobenzidine tetrachloride (Sigma).
Data analysis
NeuN- and F-J B-positive cells were counted according to our published procedure (Bae et al., 2015). Fifteen brain tissue sections were chosen in each animal with 120 μm interval. NeuN- and F-J B-positive cells in 200 × 200 μm2at the center of the CA1 stratum pyramidale were counted using an AxioM1 light microscope (Carl Zeiss) equipped with a digital camera (Axiocam, Carl Zeiss) interlinked with a PC monitor. Cell counts were analyzed as a percent, with the sham group and ischemia group (5 days) designated as 100%. To quantitatively analyze CB immunoreactivity, in brief, according to our method (Lee et al., 2016), images were calibrated into an array of 512 × 512 pixels corresponding to a tissue area of 140 × 140 μm2(40× original magnification).e mean CB immunoreactivity was determined in hippocampal CA1 pyramidal neurons by a 0–255 gray scale system in ImageJ (National Institutes of Health, MD, USA). The background density was subtracted, and the relative immunoreactivity (RI) of image file was calibrated as % using Adobe Photoshop version 8.0, and the RI was analyzed using NIH Image 1.59 soware (National Institutes of Health). RI was calibrated as %, with sham group designated as 100%.
Statistical analysis
All data are expressed as the mean ± SEM of the means across the groups and were statistically analyzed using SPSS 18.0 (SPSS, Chicago, IL, USA). Analysis of variance (ANOVA) with apost hocBonferroni’s multiple comparison test was performed to present differences among experimental groups. Statistical significance was considered atP< 0.05.
Results
CV-positive cells
CV-positive cells were obviously observed in the stratum pyramidale, which are called pyramidal neurons, in the gerbil hippocampus proper (CA1–3 area) of the sham group(Figure 1Aand1a). In the ischemic gerbils, the morphology of CV-positive pyramidal neurons in the hippocampus proper was not changed until 3 days aer ischemia/reperfusion (Figure 1C,1c,1E,1e,1G,1gand1M); however, CV-positive cells were rarely detected in the stratum pyramidale of the CA1 area, not CA2 and CA3 area from 4 days aer ischemia/reperfusion (Figure 1I,1i,1K,1kand1M).
F-J B-positive cells
F-J B-positive cells, which are dead cells, were not observed in the CA1 area of the ischemia group until 3 days post-ischemia (Figure 2A,2B,2E,2Fand2M). However, many F-J B-positive cells were observed in the pyramidal layer of the CA1 area at 4 and 5 days post-ischemia (Figure 2I,2Jand2M). F-J B-positive cells were not observed in the CA1 area of the IPC + sham group (Figure 2Cand2M). In the IPC + ischemia group, F-J B-positive cells were not observed until 3 days post-ischemia (Figure 2D,2G,2Hand2M), and 4 and 5 days aer ischemia/reperfusion, only a few F-J B-positive cells were observed in the pyramidal layer (Figure 2K,2Land2M).
CB immunoreactivity
In the sham group, CB immunoreactivity was detected in the pyramidal neurons of the CA1–3 area (Figure 3Aand3a). In the ischemia group, CB immunoreactivity was not signif icantly altered in pyramidal neurons at 1 day post-ischemia (Figure 3B,3band3m); however, CB immunoreactivity was significantly decreased only in the CA1 pyramidal neurons 2 days aer ischemia/reperfusion (Figure 3E,3eand3m), and CB immunoreactivity in the CA1 pyramidal cells was hardly detected from 3 days aer ischemia/reperfusion (Figure 3F,3I,3J,3f,3i,3jand3m). In the IPC + sham group, CB immunoreactivity in pyramidal neurons of the CA1–3 area was not different from that in the sham group (Figure 3C,4Cand4M). In the IPC + ischemia group, CB immunoreactivity in all pyramidal cells was steadily maintained until 5 days post-ischemia (Figure 3D,3G,3H,3K,3L,3d,3g,3h,3k,3land3m).
Discussion
Transient global cerebral ischemia selectively kills neurons in the hippocampus, namely, CA1 pyramidal neurons slowly die a few days aer transient global cerebral ischemia (Kirino, 1982; Kirino and Sano, 1984; Kirino, 2000). In the present study, we found a striking loss of pyramidal neurons in the pyramidal layer of the CA1 area, which are named CA1 pyramidal cells, from 4 days aer 5-minute transient ischemiaviaCV staining and F-J B histofluorescence staining. This finding is consistent with previous reports using gerbils that were subjected to 5-minute transient cerebral ischemia (Kim et al., 2014; Yan et al., 2014).
Previous studies demonstrated that IPC, which was induced by non-fatal brief transient ischemia, generated ischemic tolerance and protected neuronal damage/death following subsequent fatal transient ischemia (Kirino et al., 1996; Lehotsky et al., 2009). We investigated the neuroprotective effect of IPC against 5-minute transient cerebral ischemia in the CA1 area using CV staining and F-J B histofluorescence staining, and our finding was similar to that in previous studies (Lee et al., 2014; Kim et al., 2015). Brief (2-minute) transient ischemia in the brain does not cause the death of CA1 pyramidal neurons, and however, protects CA1 pyramidal neurons aer a subsequent longer time (5-minute) of transient cerebral ischemia in gerbils. Previous studies have demonstrated that IPC mediates neuroprotection through attenuating ubiquitin aggregation (Lee et al., 2014), reducing oxidative damage (Lee et al., 2015a; Park et al., 2016), increasing the level of anti-inflammatory cytokines (Kim et al., 2015) and inhibiting Na(+)/H(+) exchanger 1 expression (Lee et al., 2015b). However, to the best of our knowledge, there are no studies correlating CB immunoreactivity with IPC-mediated neuroprotection following subsequent transient ischemic insults.
Calcium ions conduct important physiological functions that activate and regulate the fast transport of substances in axons, membrane excitability in neurons, and neurotransmitter synthesis and release (Heizmann and Braun, 1992). However, massive neuronal degeneration takes place in several brain diseases and the expression of calcium binding proteins changes during the course of the diseases (Heizmann and Braun, 1992). In ischemic brain injury, calcium ions are overloaded and lead to the activation of biochemical processes, enzymatic breakdowns of proteins, lipids and nucleic acids, mitochondrial malfunction, energy failure, and finally the destruction of neurons (Lee et al., 1999; Li et al., 2011). In addition, Sadowski et al. (2002) reported that, in a rat model of cardiac arrest, CB immunoreactivity disappeared completely in CA1 pyramidal neurons 3 days after cardiac arrest. In our present research, CB immunoreactivity in the pyramidal cells of the CA1 area began to be significantly decreased from 2 days and hardly detected 5 days aer transient ischemia. We, in addition, reported that CA1 pyramidal cells of the young gerbils showed more resistance to transient cerebral ischemia than those in the adult gerbils and CB expression in the CA1 pyramidal cells was longer maintained in the young gerbils than in the adult gerbils (Lee et al., 2013b).

Figure 2 Fluoro-Jade B (F-J B) histofluorescence staining of the CA1 area in the sham (A), IPC + sham (C), ischemia (B, E, F, I, and J), and IPC + ischemia (D, G, H, K, and L) groups.
In this study, we found that CB expression was consistently maintained in the IPC + ischemia group, which shows IPC-mediated protection of the CA1 pyramidal cells against a fatal subsequent transient ischemia. It seems that the maintenance of CB expression in the IPC + ischemia group might be related with the protection of IPC against a fatal subsequent cerebral ischemia. It is well known that calcium binding proteins including CB have a greater capacity of intercellular calcium ion buffering that would be more resistant to some brain disorders (Heizmann and Braun, 1992). However, studies regarding CB-mediated neuroprotection in cerebral ischemic condition have been reported by some researchers. Yenari et al. (2011) injected viral vector-mediated CB into the striatum of the rat and found the attenuation of neuronal damage/death in the striatum following focal cerebral ischemia (Yenari et al., 2001). Freimann et al. (2010) overexpressed CB in the striatum and cerebral cortex in the mouse using an adeno-associated viral vector for long time and found neuroprotective effects after focal cerebral ischemia induced by the occlusion of the middle cerebral artery (Freimann et al., 2010). Sung et al. (2012) reported that ginkgo biloba extract prevented the reduction of parvalbumin, a kind of calcium binding protein, in cerebrocortical neuronal cells of the rat after transient focal cerebral ischemia. Koh (2013) reported that nicotinamide restored the reduction of parvalbumin in the rat cerebral cortex induced by transient focal cerebral ischemia.
In brief, our results showed that IPC protected pyramidal neurons of the hippocampal CA1 area against subsequent fatal transient ischemia and restored the reduction of CB expression in the pyramidal neurons in the hippocampal CA1 area aer subsequent fatal transient ischemia.ese results indicate that CB in the brain plays important roles in the neuroprotection of neurons against brain damage including ischemic insults.
Acknowledgments:We would like to thank Mr. Seung Uk Lee (Department of Physiology, College of Medicine, and Institute of Neurodegeneration and Neuroregeneration, Hallym University, Chuncheon, South Korea) for his technical help in this study.
Author contributions:All authors were responsible for design, implementation and evaluation of the study and approved the final version of this paper.
Conflicts of interest:None declared.
Research ethics:Procedures for animal handling and care complied with the guidelines that follow the current international laws and policies (Guide for the Care and Use of Laboratory Animals,e National Academies Press, 8th Ed., 2011), and all the experimental protocols of this study were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC), Kangwon National University (approval no. KW-160802-1).
Open access statement:
Contributor agreement:A statement of “Publishing Agreement” has been signed by an authorized author on behalf of all authors prior to publication.
Plagiarism check:This paper has been checked twice with duplication-checking soware ienticate.
Peer review:A double-blind and stringent peer review process has been performed to ensure the integrity, quality and significance of this paper.
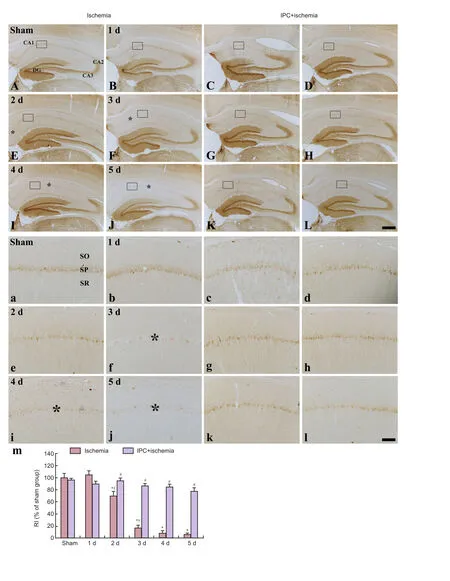
Figure 3 Low magnification of calbindin-D28k (CB) immunohistochemistry in the hippocampus of the sham (A), IPC + sham (C), ischemia (B, E, F, I, and J), and IPC + ischemia (D, G, H, K, and L) groups.
Bae EJ, Chen BH, Shin BN, Cho JH, Kim IH, Park JH, Lee JC, Tae HJ, Choi SY, Kim JD, Lee YL, Won MH, Ahn JH (2015) Comparison of immunoreactivities of calbindin-D28k, calretinin and parvalbumin in the striatum between young, adult and aged mice, rats and gerbils. Neurochem Res 40:864-872.
Baimbridge KG, Celio MR, Rogers JH (1992) Calcium-binding proteins in the nervous system. Trends Neurosci 15:303-308.
Cai F, Liu J, Li C, Wang J (2015) Intracellular calcium plays a critical role in the microcystin-LR-elicited neurotoxicity through PLC/IP3 pathway. Int J Toxicol 34:551-558.
D’Orlando C, Fellay B, Schwaller B, Salicio V, Bloc A, Gotzos V, Celio MR (2001) Calretinin and calbindin D-28k delay the onset of cell death aer excitotoxic stimulation in transfected P19 cells. Brain Res 909:145-158.
Fan Y, Shi L, Gu Y, Zhao Y, Xie J, Qiao J, Yang GY, Wang Y, Lu CZ (2007) Pretreatment with PTD-calbindin D 28k alleviates rat brain injury induced by ischemia and reperfusion. J Cereb Blood Flow Metab 27:719-728.
Freimann FB, Crome O, Shevtsova Z, Bahr M, Kugler S (2010) Evaluation of long-term upregulation of Calbindin D28K as a preventive approach for ischaemic stroke. Int J Stroke 5:319-320.
Guo Q, Christakos S, Robinson N, Mattson MP (1998) Calbindin D28k blocks the proapoptotic actions of mutant presenilin 1: reduced oxidative stress and preserved mitochondrial function. Proc Natl Acad Sci U S A 95:3227-3232.
Heizmann CW, Braun K (1992) Changes in Ca(2+)-binding proteins in human neurodegenerative disorders. Trends Neurosci 15:259-264.
Kim DW, Lee JC, Cho JH, Park JH, Ahn JH, Chen BH, Shin BN, Tae HJ, Seo JY, Kang IJ, Hong S, Kim YM, Won MH, Kim IH (2015) Neuroprotection of ischemic preconditioning is mediated by anti-inflammatory, not pro-inflammatory, cytokines in the Gerbil hippocampus induced by a subsequent lethal transient cerebral ischemia. Neurochem Res 40:1984-1995.
Kim IH, Yoo KY, Park JH, Yan BC, Ahn JH, Lee JC, Kwon HM, Kim JD, Kim YM, You SG, Kang IJ, Won MH (2014) Comparison of neuroprotective effects of extract and fractions from Agarum clathratum against experimentally induced transient cerebral ischemic damage. Pharm Biol 52:335-343.
Kirino T (1982) Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res 239:57-69.
Kirino T (2000) Delayed neuronal death. Neuropathology 20 Suppl:S95-97.
Kirino T, Sano K (1984) Selective vulnerability in the gerbil hippocampus following transient ischemia. Acta Neuropathol 62:201-208.
Kirino T, Nakagomi T, Kanemitsu H, Tamura A (1996) Ischemic tolerance. Adv Neurol 71:505-511.
Klapstein GJ, Vietla S, Lieberman DN, Gray PA, Airaksinen MS,oenen H, Meyer M, Modyi(1998) Calbindin-D28k fails to protect hippocampal neurons against ischemia in spite of its cytoplasmic calcium buffering properties: evidence from calbindin-D28k knockout mice. Neuroscience 85:361-373.
Koh PO (2013) Nicotinamide restores the reduction of parvalbumin in cerebral ischemic injury. J Vet Med Sci 75:225-229.
Kovalska M, Kovalska L, Mikuskova K, Adamkov M, Tatarkova Z, Lehotsky J (2014) p-ERK involvement in the neuroprotection exerted by ischemic preconditioning in rat hippocampus subjected to four vessel occlusion. J Physiol Pharmacol 65:767-776.
Lee JC, Ahn JH, Lee DH, Yan BC, Park JH, Kim IH, Cho GS, Kim YM, Lee B, Park CW, Cho JH, Lee HY, Won MH (2013a) Neuronal damage and gliosis in the somatosensory cortex induced by various durations of transient cerebral ischemia in gerbils. Brain Res 1510:78-88.
Lee JC, Kim IH, Cho GS, Park JH, Ahn JH, Yan BC, Kwon HM, Kim YM, Cheon SH, Cho JH, Lee HY, Won MH, Seo JY (2014) Ischemic preconditioning-induced neuroprotection against transient cerebral ischemic damage via attenuating ubiquitin aggregation. J Neurol Sci 336:74-82.
Lee JC, Kim IH, Park JH, Ahn JH, Cho JH, Cho GS, Tae HJ, Chen BH, Yan BC, Yoo KY, Choi JH, Lee CH, Hwang IK, Kwon YG, Kim YM, Won MH (2015a) Ischemic preconditioning protects hippocampal pyramidal neurons from transient ischemic injury via the attenuation of oxidative damage through upregulating heme oxygenase-1. Free Radic Biol Med 79:78-90.
Lee JC, Cho JH, Kim IH, Ahn JH, Park JH, Cho GS, Chen BH, Shin BN, Tae HJ, Park SM, Ahn JY, Kim DW, Bae EJ, Yong JH, Kim YM, Won MH, Lee YL (2015b) Ischemic preconditioning inhibits expression of Na(+)/H(+) exchanger 1 (NHE1) in the gerbil hippocampal CA1 region aer transient forebrain ischemia. J Neurol Sci 351:146-153.
Lee JC, Tae HJ, Kim IH, Cho JH, Lee TK, Park JH, Ahn JH, Choi SY, Bai HC, Shin BN, Cho GS, Kim DW, Kang IJ, Kwon YG, Kim YM, Won MH, Bae EJ (2016) Roles of HIF-1alpha, VEGF, and NF-kappaB in ischemic preconditioning-mediated neuroprotection of hippocampal CA1 pyramidal neurons against a subsequent transient cerebral ischemia. Mol Neurobiol doi:10.1007/s12035-016-0219-2.
Lee JM, Zipfel GJ, Choi DW (1999) The changing landscape of ischaemic brain injury mechanisms. Nature 399:A7-14.
Lee YJ, Yan BC, Park JH, Ahn JH, Kim IH, Lee JC, Lee HY, Kim YM, Won MH, Cho JH (2013b) Differences of calcium binding proteins immunoreactivities in the young hippocampal CA1 region from the adult following transient ischemic damage. J Neurol Sci 326:40-47.
Lehotsky J, Burda J, Danielisova V, Gottlieb M, Kaplan P, Saniova B (2009) Ischemic tolerance: the mechanisms of neuroprotective strategy. Anat Rec (Hoboken) 292:2002-2012.
Li MH, Inoue K, Si HF, Xiong ZG (2011) Calcium-permeable ion channels involved in glutamate receptor-independent ischemic brain injury. Acta Pharmacol Sin 32:734-740.
Liu XQ, Sheng R, Qin ZH (2009)e neuroprotective mechanism of brain ischemic preconditioning. Acta Pharmacol Sin 30:1071-1080.
Ohk TG, Yoo KY, Park SM, Shin BN, Kim IH, Park JH, Ahn HC, Lee YJ, Kim MJ, Kim TY, Won MH, Cho JH (2012) Neuronal damage using fluoro-jade B histofluorescence and gliosis in the striatum aer various durations of transient cerebral ischemia in gerbils. Neurochem Res 37:826-834.
Park SM, Park CW, Lee TK, Cho JH, Park JH, Lee JC, Chen BH, Shin BN, Ahn JH, Tae HJ, Shin MC, Ohk TG, Won MH, Choi SY, Kim IH (2016) Effect of ischemic preconditioning on antioxidant status in the gerbil hippocampal CA1 region aer transient forebrain ischemia. Neural Regen Res 11:1081-1089.
Saad MA, Abdelsalam RM, Kenawy SA, Attia AS (2015) Ischemic preconditioning and postconditioning alleviates hippocampal tissue damage through abrogation of apoptosis modulated by oxidative stress and inflammation during transient global cerebral ischemia-reperfusion in rats. Chem Biol Interact 232:21-29.
Sadowski M, Lazarewicz JW, Jakubowska-Sadowska K, Wisniewski HM, Mossakowski MJ, Brown WT (2002) Long-term changes in calbindin D(28K) immunoreactivity in the rat hippocampus after cardiac arrest. Neurosci Lett 321:90-94.
Sung JH, Shah FA, Cho EH, Gim SA, Jeon SJ, Kim KM, Kim YM, Kim MO, Koh PO (2012) Ginkgo biloba extract (EGb 761) prevents the ischemic brain injury-induced decrease in parvalbumin expression. Lab Anim Res 28:77-82.
Verdaguer E, Brox S, Petrov D, Olloquequi J, Romero R, de Lemos ML, Camins A, Auladell C (2015) Vulnerability of calbindin, calretinin and parvalbumin in a transgenic/knock-in APPswe/PS1dE9 mouse model of Alzheimer disease together with disruption of hippocampal neurogenesis. Exp Gerontol 69:176-188.
Yan BC, Ohk TG, Ahn JH, Park JH, Chen BH, Lee JC, Lee CH, Shin MC, Hwang IK, Moon SM, Cho JH, Won MH (2014) Differences in neuronal damage and gliosis in the hippocampus between young and adult gerbils induced by long duration of transient cerebral ischemia. J Neurol Sci 337:129-136.
Yenari MA, Minami M, Sun GH, Meier TJ, Kunis DM, McLaughlin JR, Ho DY, Sapolsky RM, Steinberg GK (2001) Calbindin d28k overexpression protects striatal neurons from transient focal cerebral ischemia. Stroke 32:1028-1035.
Copyedited by Li CH, Song LP, Zhao M
How to cite this article: Kim IH, Jeon YH, Lee TK, Cho JH, Lee JC, Park JH, Ahn JH, Shin BN, Kim YH, Hong S, Yan BC, Won MH, Lee YL (2017) Neuroprotective eects of ischemic preconditioning on hippocampal CA1 pyramidal neurons through maintaining calbindin D28k immunoreactivity following subsequent transient cerebral ischemia. Neural Regen Res 12(6):918-924.
*Correspondence to:
Moo-Ho Won, D.V.M., Ph.D., or Yun Lyul Lee, D.V.M., Ph.D., mhwon@kangwon.ac.kr or yylee@hallym.ac.kr.
orcid:
0000-0002-7178-6501 (Moo-Ho Won) 0000-0003-0504-5825 (Yun Lyul Lee)
10.4103/1673-5374.208573
Accepted: 2017-05-15
- 中國神經(jīng)再生研究(英文版)的其它文章
- Synaptosomal-associated protein 25 may be an intervention target for improving sensory and locomotor functions after spinal cord contusion
- On the role of endogenous neurotoxins and neuroprotection in Parkinson’s disease
- Interfacing peripheral nerve with macro-sieve electrodes following spinal cord injury
- Neuroprotective effects of ganoderma lucidum polysaccharides against oxidative stress-induced neuronal apoptosis
- Mechanisms underlying the promotion of functional recovery by deferoxamine after spinal cord injury in rats
- Galantamine protects against beta amyloid peptide-induced DNA damage in a model for Alzheimer’s disease