
Identification and characterization of short tandem repeats in the Tibetan macaque genome based on resequencing data
2018-06-27 09:03:20SanXuLiuWeiHouXueYanZhangChangJunPengBiSongYueZhenXinFanJingLi
Zoological Research 2018年4期
San-Xu Liu,Wei Hou,Xue-Yan Zhang,Chang-Jun Peng,Bi-Song Yue,2,Zhen-Xin Fan,Jing Li,2,*
1Key Laboratory of Bio-Resource and Eco-Environment of Ministry of Education,College of Life Sciences,Sichuan University,Chengdu
Sichuan 610065,China
2Sichuan Key Laboratory of Conservation Biology on Endangered Wildlife,College of Life Sciences,Sichuan University,Chengdu Sichuan 610065,China
INTRODUCTION
Short tandem repeats(STRs),also known as microsatellites,are highly variable repetitive elements with nucleotide motifs of 1–6 bp and are ubiquitous in most eukaryotic organisms(Oliveira et al.,2006).Microsatellite mutations are generally derived from replication slippage,leading to the insertion or deletion of one or several repeat motifs(Levinson&Gutman,1987;Schl?tterer,2000;Schl?tterer&Tautz,1992).STRs located in both the coding and non-coding areas of DNA can affect gene expression and transcription(Chistiakov et al.,2006;Li et al.,2004),especially STRs in the coding regions.For example,certain human neurological disorders are caused by trinucleotide repeat expansions in coding regions(La Spada et al.,1991;MacDonald et al.,1993). Previous studies have also indicated that the mutation processes of STR loci can vary,even among closely related species,resulting in interspecific differences in STR allele size(Rubinsztein et al.,1995;Schl?tterer,1998;Webster et al.,2002).STRs are frequently applied to genetic mapping,forensic identification,and phylogenetic studies as molecular markers due to their high abundance,neutrality,high variability,and codominance(Schl?tterer,1998).Recent studies have also emphasized the role of STRs in the genetic architecture of quantitative human traits(Gymrek et al.,2016).In addition,STRs have been used as molecular markers for the study of genetic diversity and population genetic structure(Qin et al.,2016;Sunnucks,2000).Here,we determined genome-wide STR variation in Tibetan macaque populations based on high-throughput sequencing.
The Tibetan macaque(Macaca thibetana)is one of 23 extant species of the genusMacaca(Primates:Cercopithecidae)and is endemic to China.Its geographic distribution extends from southwest to east regions in China (Jiang et al.,1996). Jiang et al. (1996)dividedM.thibetanainto four subspecies according to variations in external morphology,hair color,cranial features,and geographical distribution:namely,Macaca thibetana thibetana,Macaca thibetana pullus,Macaca thibetana huangshanensis,andMacaca thibetana guizhouensis.The species is an increasingly studied animal model in biomedical research due to its ease of domestication,large body size,long life span,and physiological characteristics analogous to those of humans(Yi et al.,2012).Studies on intraocular pressure(Liu et al.,2011)and liver transplantation(Ji et al.,2015)are two good application examples.However,habitat destruction,deforestation,and illegal poaching have led to considerable fragmentation of naturalMacacapopulations,with a resulting decrease in wild Tibetan macaque populations observed in recent years(Jiang et al.,2016).Currently,the Tibetan macaque is a vulnerable and endangered species(Jiang et al.,2016),and is classified as a Category II species under the Chinese Wild Animal Protection Law.Thus,studies on its genetic variability and diversity are urgently needed to assess genetic differentiation and develop effective conservation strategies. To date,previous studies have recognized 56 STR markers in Tibetan macaques by genomic library construction and cross-species amplification(Jia et al.,2011,2012;Li et al.,2014;Yang et al.,2017).Nevertheless,the numbers of identified STR markers are insufficient for accurate estimation,conservation,and management of Tibetan macaques. Most STRs in the Tibetan macaque genome remain unidentified and there is no information available on the comparative analysis of whole genome STRs due to the lack of genomic data.
Whole genome pro files of STR variations in non-model organisms have not yet been conducted,despite the discovery of STRs in the 1980s(Guichoux et al.,2011).This is due to the associated expense as well as the inefficient and laborious construction of genomic libraries and enrichment of clones containing STR motifs(Castagnone-Sereno et al.,2010;Powell et al.,1996).Recently,the advent of high-throughput sequencing technology has produced considerable genomic data,providing the opportunity for genome-wide analysis of STR variations. To date,STR marker development and polymorphism analyses based on high-throughput sequencing data have been performed in humans(Willems et al.,2014),cattle(Xu et al.,2016),maize(Qu&Liu,2013),pig(Liu et al.,2017a),faba bean(Abuzayed et al.,2017),andAngelica gigas Nakai(Gil et al.,2017).However,the distribution and variation of genome-wide STRs in the Tibetan macaque remain largely unknown.To identify and characterize all potential STR loci and clarify the differences in STRs among Tibetan macaque individuals,we systematically profiled STR distribution in the Tibetan macaque genome and STR variations between Emei,Jianyang,and Huangshan individuals based on whole genome re sequencing data.These profiling results will contribute to our understanding of Tibetan macaque species variability.
MATERIALS AND METHODS
Genomic samples and high-throughput sequencing
Five Tibetan macaques,two obtained from Huangshan Mountain(Anhui Province,HS)and three captured from Emei Mountain(one individual)and Jianyang city(two individuals)in Sichuan Province(SC),China,were used for whole genome resequencing. Genomic DNA was isolated from blood and muscle tissue samples(Supplementary Table S1).All procedures were approved by the local Ethics Review Board and were in accordance with all relevant national and international regulations. We generated 150-bp paired-end reads with insertion sizes of approximately 350 bp on the Illumina HiSeq X Ten platform.In total,93.68–104.19 Gb of clean data were obtained after filtering out low-quality reads using the NGS QC toolkit with a quality score of 20 and percentage of read length less than 75%of given quality(Patel&Jain,2012)at a sequencing depth of over 30×in the five samples,respectively.The resequencing genome data were deposited in the GSA dataset(http://bigd.big.ac.cn/gsa/) under accession number CRA000789.
Genome assembly and characterization of STRs
The high-quality clean reads of one Emei Mountain macaque were assembled using the short reads assembling program ABySS 2.0(Simpson et al.,2009).The resulting short contigs(<150 bases)were excluded from the assembly.We then used MSDB v2.4(Du et al.,2013)to search STRs using the same settings and parameters as described previously(Liu et al.,2017b).
Genotyping of STRs based on resequencing data
Genotyping of the STRs was performed using lobSTR software(version 4.0.6),a rapid and accurate algorithm for STR pro filing based on whole-genome sequencing data(Gymrek et al.,2012).The algorithm can avoid gapped alignment and address specific noise patterns in STR calling.In this study,a lobSTR reference was first constructed based on rhesus macaque(GCF_000772875.2)STR data(generated in Liu et al.,2017b)using lobstr_index.py script.The resequencing data were then aligned to the rhesus macaque reference genome with default parameters.The output BAM files were sorted with SAMtools.Finally,we analyzed STR allelotypes based on the alignment files of the five samples.In total,we obtained 840356 STRs across the five macaque individuals after removing low-quality STRs(QUAL<30),which were used for downstream analyses.Statistical analyses were performed using SPSS version 19.
Genetic relatedness analysis of STRs
Tetranucleotide STR loci from all five samples were employed for genetic analysis. Each STR allele different from the reference allele size was coded as a binary string of all zeros,and the remaining alleles were set to one.Genetic distance was calculated using the vegan package in the R program(version 3.0.2)(Team,2000).Finally,we constructed a phylogenetic tree based on the neighbor-joining method in MEGA 5.2(Hall,2013).
Screening of polymorphic STRs
Allele numbers of each STR loci were counted based on genotyping data,and high-quality,polymorphic tetranucleotide loci were screened using VCFtools by the application of“--min-alleles 4”and “--maf 0.1”parameters.Ten loci from these STRs were randomly selected to validate efficiency and polymorphism in the 16 Tibetan macaque samples by agarose gel electrophoresis and capillary electrophoresis. Cervus software was employed to calculate the observed number of alleles(Na),mean observed heterozygosity(Ho),mean expected heterozygosity(He),and polymorphic information content(PIC).
RESULTS
Sequencing,assembly,and STR overview of the Tibetan macaque genome
We resequenced the genomes of five Tibetan macaques from Sichuan and Huangshan populations using Illumina sequencing technology,generating over 30-fold sequencing depth for each individual. After data filtering,we obtained a total of 312252249 clean sequencing reads with 150-bp and 350-bp insert fragments,corresponding to 93.68–95.45 G base pairs with GC content of approximately 45%and over 94.06%Q20 bases(base quality more than 20)in each sample(Supplementary Table S1).In the assembled genome,we acquired a total of 1223752 contigs with a total length of 2.66 Gb and N50 of 4966 bp.The average GC content of the genomic sequences was 40.48%.The maximum and average lengths of the contigs were 97085 bp and 2172.38 bp,respectively(Table 1).The length distributions of the contigs can be seen in Supplementary Figure S1.

Table 1 Assembly results for the M.thibetana genome
Hereafter,we analyzed the distribution of STRs in the assembly using MSDB2.4 software.All 1223752 contigs generated in this study were used to identify potential STRs with minimum repeats of 12,7,5,4,4,and 4 for all motifs from mono-to hexa-nucleotide STRs,respectively.As expected,perfect STRs were the most abundant category,followed by compound STRs,interrupted compound and interrupted perfect STRs,and complex STRs(Supplementary Table S2).A total of 1077790 perfect STRs(accounting for approximately 0.78%of the assembly)were mined in 521523 contigs,with 252041 sequences containing more than one STR(Table 1).The frequency,average length,and GC content of the potential STRs were also analyzed. Among the 1077790 STRs,mononucleotide repeats with the highest frequency of 234.7 loci/Mb were the most abundant types,accounting for 57.89%of the total number of STRs,followed by tetra-(181344 loci,16.83%)and di-nucleotide(165769 loci,15.38%)repeats,with hexanucleotide(6347 loci,0.59%)repeats the least abundant(Table 2).Average length of the STR core sequences increased with repeat motifs from mono-to hexa-nucleotide STRs,except for trinucleotide repeats,with the total average length found to be 19.19 bp. Analysis of GC content for the six STR types showed that GC content was highest in dinucleotide STRs,accounting for 48.75%of dinucleotide STRs in the assembly,followed by tri-,hexa-and tetra-nucleotide repeats.The lowest GC-content was found in the mononucleotide STRs,accounting for 0.48%in the genome(Table 2).
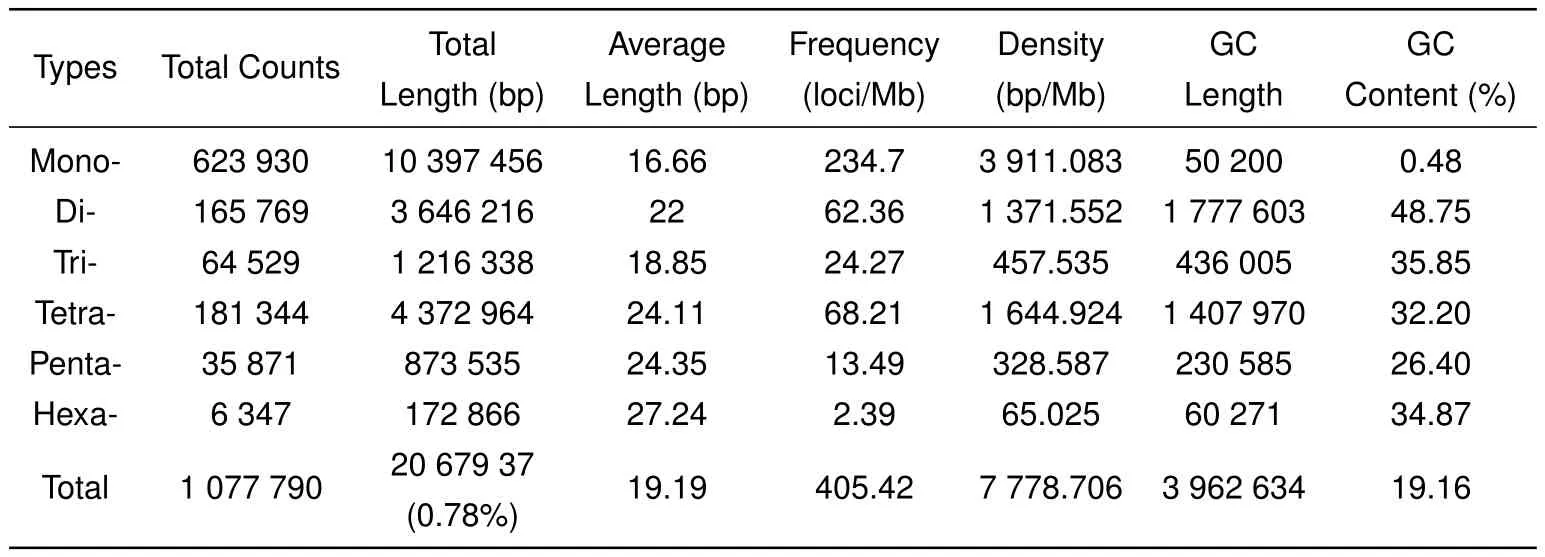
Table 2 Number,length,frequency,density,and GC content of perfect STRs
We also found that most repeat motifs were AT-rich,except for the dinucleotide repeats.With a frequency of 233.81 loci/Mb,the A/T mononucleotide repeat was the most frequent motif in the STRs,followed by the AC/GT(42.6 loci/Mb),AG/CT(17.09 loci/Mb),AAAC/GTTT(13.53 loci/Mb),ATTT/AAAT(11.76 loci/Mb),AAGG/CCTT(11.7 loci/Mb),AAAG/CTTT(11.35 loci/Mb),AAC/GTT(8.47 loci/Mb),GTTTT/AAAAC(6.22 loci/Mb),and ATT/AAT(5.28 loci/Mb)motifs.The frequency of the remaining motifs was 36.52 loci/Mb(Figure 1).The number of major repeats ranged from 12 to 40 for mono-,7 to 27 for di-,5 to 20 for tri-,4 to 15 for tetra-,4 to 12 for penta-,and 4 to 8 for hexa-nucleotide repeat,respectively(Figure 2).These loci accounted for 97.89%of the total counts of each STR type.
Alignment and genotyping of STRs
To explore differences in STR distribution among the Tibetan macaques,we usedlob STR software to examine the high-throughput sequencing data of the five individuals from the Huangshan and Sichuan populations(Supplementary Table S1). A total of 1060419 STRs were found,with the 1294455 perfect STRs located in the rhesus macaque chromosomes used as a reference.After removing low-quality STRs(QUAL<30),we achieved a total of 840356 STRs across the five macaques.On average,we obtained genotypes for 713685.8 STR loci,with a mean coverage of 5.94-fold per individual,of which 354834.2 STR loci(49.72%)had 5-fold greater call coverage(Figure 3A).Further observation showed that 526699 STR loci(62.68%)of the 840356 STRs called in our study were shared by the five macaques and 35352 STR loci(4.20%)were unique to one individual.In addition,63124,87390,and 127791 STRs were identified in two,three,and four individuals,respectively(Figure 3A).The STR loci showed greatest alignment to chr 1,followed by chr 2,chr3,chr 7,and chr 5,and finally chr Y(Figure 3B).Comparison of allele number of di-and tri-nucleotide STR motifs indicated that AT repeats had the highest proportion in dinucleotide STR motifs with greater than one allele,followed by AC,AG,and CG repeats. For trinucleotide STR motifs with more than one allele,AAG,AAT,and AAC were found at higher proportions.Conversely,for STR motifs with only one allele(no polymorphism),the proportions of CG and CCG were higher than those of the other motif types(Figure 3C).

Figure 1 Distribution of STR motifs in M.thibetana

Figure 2 Distribution of STR types in the M.thibetana genome by repeat time
Screening and initial validation of polymorphic STRs
Analysis of the allele number of STRs among the five individuals indicated that loci with only one allele were underrepresented in all individuals,accounting for 1.41%(11869 loci)of all loci.Among these loci,trinucleotide STRs had the highest proportion,accounting for 6.03%(1726 loci)of the total number of trinucleotide STRs;and mononucleotide STRs were relatively underrepresented,accounting for 0.54%(3205 loci)of mononucleotide STRs.STR loci with two alleles accounted for the highest proportion in di-to hexa-nucleotide STRs. The proportion of STRs with two alleles(22.93%,31.51%,55.88%,52.12%,58.09%,and 58.15%from monoto hexa-nucleotide STRs,respectively)increased with motif length. Loci with more than four alleles(pSTRs)had the highest proportion in mono-(245673 loci,41.56%)and di-nucleotide loci(52180 loci,40.42%),followed by tetra-(13307 loci,18.84%),penta-(2560 loci,14.50%),hexa-(456 loci,13.85%),and tri-nucleotide STRs(3771 loci,13.17%)(Figure 3D).pSTRs accounted for 37.83%of the total STRs.
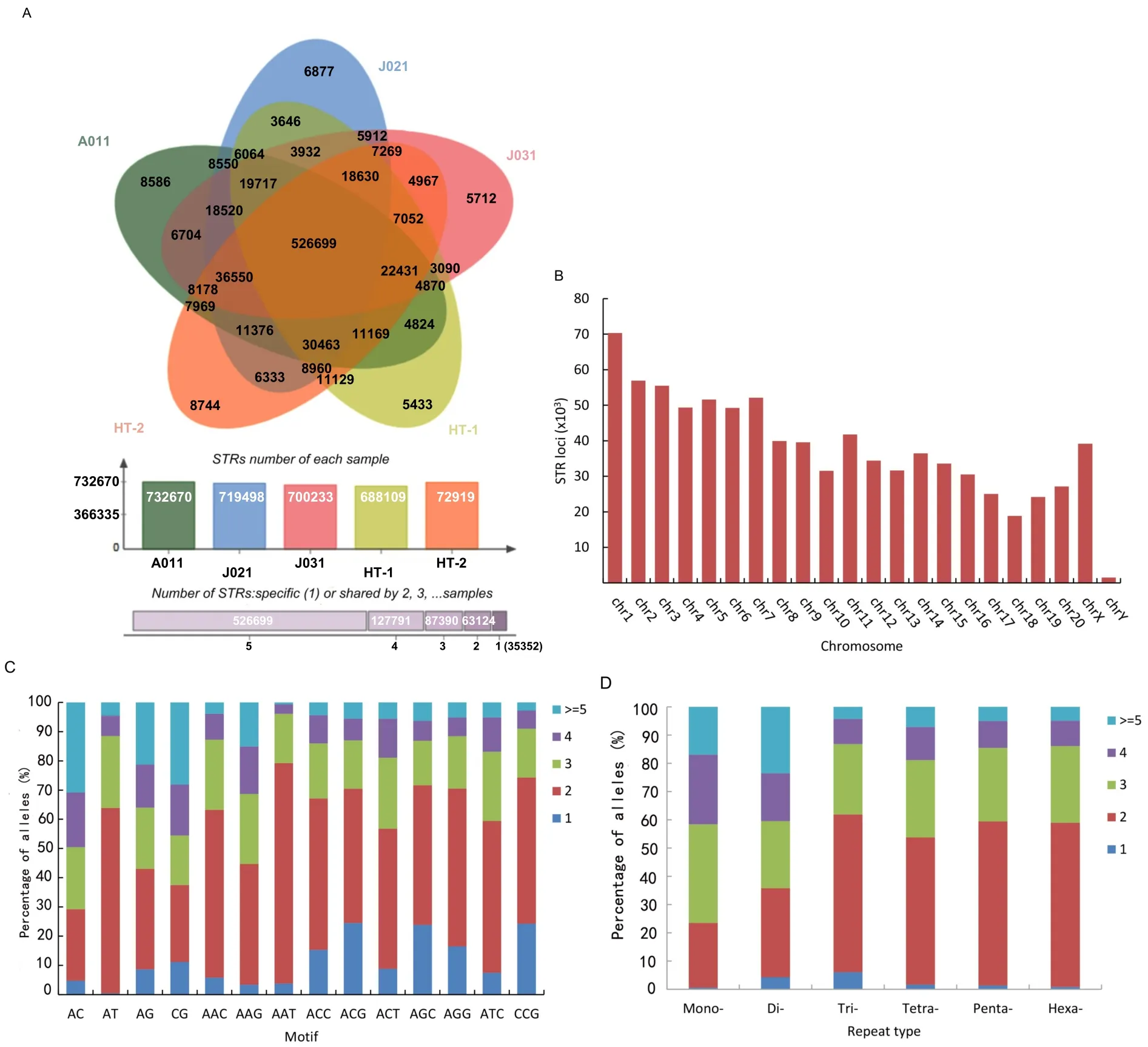
Figure 3 Alignment results of STRs
We also characterized 3325 tetranucleotide STRs that showed at least four alleles with a frequency of over 0.1.The coordinate information of these loci can be seen in Supplementary Table S3.Ten loci from these STRs were randomly selected to validate efficiency and polymorphism.Among them,the forward primers of six STR loci that were amplified successfully in the 16 samples were labeled by fluorescence(FAM/HEX).Finally,four to seven alleles were detected across each locus of the six loci. TheHo andHe values ranged from 0.40 to 0.88(mean=0.55)and 0.58 to 0.81(mean=0.69),respectively,and thePICranged from 0.519 to 0.754(mean=0.625)for each locus(Table 3).Primer information of the six markers is shown in Supplementary Table S4.

Table 3 Number,length,frequency,density,and GC content of perfect STRs
Variation and genetic relatedness analysis based on genotyping data of STRs
To obtain more accurate and reliable results,we focused on the 526699 loci called in all five macaques,including 354577 mono-,96370 di-,20271 tri-,41166 tetra-,12042 penta-,and 2273 hexa-nucleotide loci. We divided these loci into four allelotype categories:homozygous reference where both alleles are aligned to the reference;heterozygous reference where one allele is aligned to the reference;homozygous nonreference where both alleles are not mapped to the reference but are the same;and heterozygous nonreference where both alleles are different and do not match the reference.For mononucleotide repeats,as shown in Figure 4 and Supplementary Table S5,the proportion of homozygous reference/nonreference loci was lower in SC than in HS,whereas,the proportion of heterozygous reference/nonreference was higher in SC than in HS.The distributional pattern of the four allelotype categories in the other repeat types was similar to that in the mononucleotide repeats(Figure 4 and Supplementary Table S5).These results showed that Tibetan macaques in HS had a higher proportion of homozygous reference and nonreference loci than that in SC for each repeat type.Conversely,SC individuals had more heterozygous reference/nonreference loci(Figure 4).Overall,there was a far higher number of homozygous STRs than heterozygous STRs in all five Tibetan macaques,and the difference was significant(P<0.05,t-test).The percentage of homozygous loci was positively correlated with the length of the repeat motif(Figure 4).Compared with the reference alleles,the proportion of base pair deletions in the STRs was greater than that of insertions for the Tibetan macaques(P<0.05,t-test),and the proportion of insertions and mean variations in STR allele size were slightly larger in SC than in HS(Supplementary Table S6). On average,these loci demonstrated a 3.4 bp difference,with differences greater than 5 bp accounting for approximately 18.30%in each individual.Furthermore,insertions accounted for 30.60%of loci and base pair deletions accounted for 49.83%of loci(Figure 5 and Supplementary Table S6).
To measure genetic relatedness,41166 tetranucleotide STR locicalled in all five macaques were used for relatedness analysis. The phylogenetic tree constructed by neighbor-joining based on genotyping data of the tetranucleotide loci showed that the three individuals from SC were clustered onto one branch and the two individuals from HS were clustered onto another branch.On the SC branch,the two individuals from Jianyang and one individual from EM showed little genetic differentiation(Supplementary Figure S2).This result was consistent with their geographical distribution.
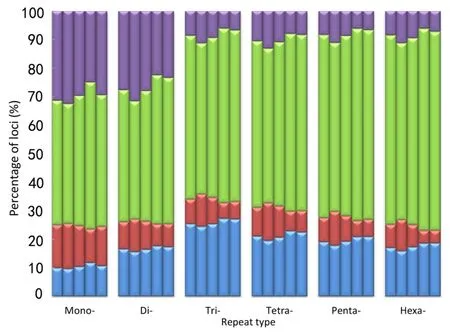
Figure 4 Comparison of the four allelotype categories for each repeat type among the five Tibetan macaques
DISCUSSION
Genome assembly and STR distribution of M.thibetana
Recently,with the development of next-generation sequencing technology and advancementin bioinformatics analysis,considerable research on genetic variation in primates has been conducted based on genome-wide sequencing,including on humans(1000 Genomes Project Consortium et al.,2015),rhesus macaques(Liu et al.,2017b;Xue et al.,2016;Zhong et al.,2016),and cynomolgus macaques(Osada et al.,2015). To date,however,few genome-scale studies have been reported in Tibetan macaques,except for the previous identification of 11.9 million single nucleotide variants(SNVs)(Fan et al.,2014). Sequencing technology now offers an unprecedented opportunity to examine STR loci of good quality and STR variations among Tibetan macaques.In the present study,based on resequencing data,STR variations and characterizations were investigated at the genome-level in Tibetan macaques.These data provide a novel genomic resource forM.thibetanaspecies and enrich the database on genetic variants in macaques.
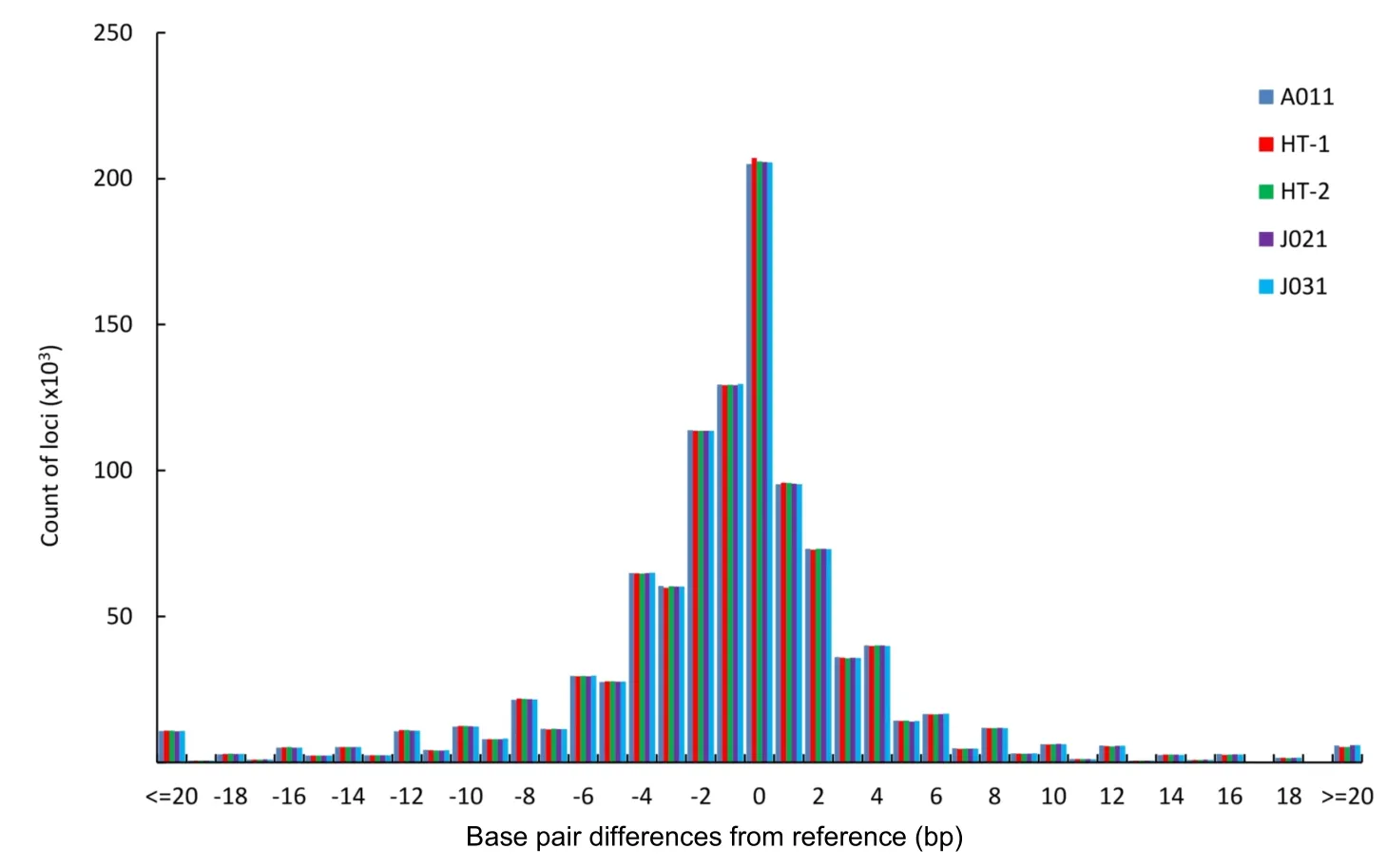
Figure 5 Distribution of allele size differences in STRs from the reference for the five M.thibetana individuals
The identification and development of Tibetan macaque STR molecular markers reported in previous studies were based on the construction of genomic libraries(Jia et al.,2011,2012;Li et al.,2014),which is both labor intensive and time consuming.Here,we employed resequencing data generated on the Illumina platform to characterize the distributional pattern of STRs. Analysis of the genome showed an average GC content of 40.48%,similar to that reported in other macaque genomes(Zimin et al.,2014).The N50 and mean length of the assembled genome were 4966 bp and 2172.38 bp,respectively,sufficient for detecting STR loci.We found that the proportion of STRs in the assembled genome was slightly lower than the 0.83%–0.88%obtained in other macaques(Liu et al.,2017b).This may be because short contigs of less than 150 bases were deleted from the assembly to obtain more reliable and accurate STR loci,leading to an incomplete genome.Mononucleotide STR loci are the most abundant repeat type in most organisms(Sharma et al.,2007),as was also found in our study.Dinucleotide and tetranucleotide STRs were the second most abundant STRs and showed similar frequencies,which is in accordance with that observed in other macaques(Liu et al.,2017b).Furthermore,analysis of GC content and repeat times for each repeat type also showed consistent results with previous study(Liu et al.,2017b).These similarities are a good explanation for the relatively high success rate of cross amplification among macaques(Engelhardt et al.,2017).We discovered that the predominant STR loci contained more A or T bases,except for dinucleotide repeats in which the AC motif was the most abundant,which has also been observed in humans(Subramanian et al.,2003).This could be due to a reported tendency that the poly(A)stretches in the genome might be more biased toward AT base pairs during STR evolution and GC-rich genome sequences are more easily mutated to produce A-rich repeats than AT-rich regions(International Human Genome Sequencing Consortium,2001;Subramanian et al.,2003).
Characterization and polymorphism of STRs based on genotyping data
Within the 840356 potential STRs identified,62.68%of loci were called in all five individuals and 4.20%of loci were only present in one individual. This may be due to the low sequencing coverage,besides true differences among different individuals.Notably,however,STRs mapped to the chromosomes exhibited similar distributional regularity with the distribution of STRs observed in other macaque chromosomes(Liu et al.,2017b).For instance,most STR loci were aligned to chr 1.Several studies have indicated a positive correlation between chromosome size and STR number(Liu et al.,2017b;Qi et al.,2015).Thus,the distribution of chromosome size inM.thibetanamay be similar to that of the rhesus macaque.In addition,we noted that GC-rich STR motifs showed lower polymorphism,which may be due to the correlation between GC-rich sequences and functionality(Benjamini&Speed,2012).
Analysis of allele number of STR loci showed that only 1.41% of the loci showed no variation among the five individuals.A similar occurrence has been reported in cattle genome research(Xu et al.,2016).Here,loci with only one allele were found at highest proportion among trinucleotide STRs compared with other STRs. Furthermore,pSTRs showed the lowest proportion among tri-and hexa-nucleotide STRs.These two results showed that variation in tri-and hexa-nucleotide STRs was smaller than that in the other four STR types.This could be attributed to the fact that these two STR types are more frequent in exons(Qi et al.,2016),which are related to gene function(La Spada et al.,1991;MacDonald et al.,1993). However,pSTRs were overrepresented in general,indicating that high polymorphic STRs were relatively abundant in the Tibetan macaque genome.These loci were found at the highest proportion in mono-and di-nucleotide STRs,followed by tetra-nucleotide STRs;however,mono-and di-nucleotide STRs are unstable and more easily produce errors in experiments(Morin et al.,2001;Taberlet et al.,1999).We also found the number of tetranucleotide STRs was higher than that of tri-,penta-,and hexa-nucleotide STRs.Thus,it is more efficient to screen polymorphic loci from tetranucleotide STRs.
From the 3325 polymorphic tetranucleotide STRs(≥4 alleles)with allele frequencies over 0.1 detected in this study,six loci were used for genetic diversity analysis of the 16M.thibetanasamples. The allele number,Ho,He,andPICresults revealed high genetic diversity within Tibetan macaques.Recent study reported 12 polymorphic loci inM.thibetanascreened from 30 STR sites in humans(Yang et al.,2017),though the amplification efficiency was lower than these STR loci identified in our study.This result indicated that the polymorphic STR loci identified in the present study can be used for amplification in theM.thibetanagenome and polymorphisms of STRs were relatively high. The combination of STR loci searched in the assembled genome and polymorphic STRs identified in this study for isolation of STRs was both efficient and time-conserving.
STR variation among five Tibetan macaque individuals
Based on the 526699 STR loci called in all five samples,we found Tibetan macaques in SC had more heterozygous reference/nonreference loci than those in HS.This indicated that genetic diversity in Tibetan macaques was higher in SC than in HS.Previous studies based on mitochondrial DNA suggest that overall genetic diversity inM.thibetanais high,and that genetic variation is higher in the SC population than in the HS population(Li et al.,2008;Zhong et al.,2013).Our results are consistent with these conclusions.Analysis of SNV distributions in the Tibetan macaque revealed that there was a higher number of homozygous SNVs than heterozygous SNVs(Fan et al.,2014). A similar occurrence was also found in the distribution of STRs,namely the majority of STRs were homozygous loci in the Tibetan macaque(P<0.05,t-test).Compared with the rhesus macaque reference alleles,the proportion of base pair deletions in the Tibetan macaque was greater than that of insertions(P<0.05,t-test),consistent with the small indels genotyped with the Genome Analysis Toolkit(GATK)(Fan et al.,2014). The average length of loci was 19.19 bp in the present assembly,which is slightly shorter than the 20.14 bp forM.mulatta(Liu et al.,2017b).These results indicate that a large proportion of STR loci may have longer repeats in rhesus macaques than orthologous loci inM.thibetana. This has also been reported from human-chimpanzee genomic sequence alignments(Webster et al.,2002).The STR loci in each Tibetan macaque showed an average difference of 3.40 bp from the rhesus macaque.The proportion of insertions and mean variations in STR allele size were slightly larger in the Emei and Jianyang individuals than in the Huangshan individuals,which may reveal differences in allele size of STRs between the two populations.Further studies are needed to examine the effect of these potential STR variations.
Genetic relationship analyses of microsatellites
The neighbor-joining tree constructed based on tetranucleotide STRs classified the five individuals into two different branches according to their geographic origin.Little genetic differentiation was observed between the Jianyang and Emei individuals.Tibetan macaques from Sichuan and Huangshan were classified into two different subspecies(M.thibetana thibetanaandM.thibetana huangshanensis)based on their external morphological and anatomical variations(Jiang et al.,1996),and the neighbor-joining tree confirmed the genetic differentiation between the two populations.Previous study based on mitochondrial DNA has demonstrated that the two populations exhibit significant genetic differentiation(Zhong et al.,2013),which may be the result of geographical barriers.Specifically,gene flow between the two populations is obstructed,leading to genetic differentiation(Zhong et al.,2013).Owing to the limited number of samples,our results provide an initial understanding of the genetic variation of STRs in Tibetan macaques,and future studies are needed to investigate population genetic variations using more samples from distinct populations.
CONCLUSIONS
In summary,we performed STR characterization in the Tibetan macaque genome and provided a genome-wide atlas of microsatellite distribution with next-generation sequencing data.The polymorphic STR loci identified from the reference genome exhibited good amplification efficiency and can be well used for population genetics.This profiling will be conducive for future genome-wide genetic analyses of the Tibetan macaque at the population scale.Analysis of genome-wide STRs also preliminarily revealed variation in the genetic diversity among Tibetan macaque populations.This study contributes to our further recognition of the genetic variation among Tibetan macaque species.
COMPETING INTERESTS
The authors declare that they have no competing interests
AUTHORS’CONTRIBUTIONS
J.L.and S.X.L.conceived and designed the study. W.H.and X.Y.Z.performed the molecular lab work.S.X.L.,X.Y.Z.,C.J.P.,and Z.X.F.analyzed the data.S.X.L.wrote the paper.J.L.and W.H.revised the paper.B.S.Y.participated in the design of the study and provided an experimental platform.All authors read and approved the final version of the manuscript.
REFERENCES
1000 Genomes Project Consortium,Auton A,Brooks LD,Durbin RM,Garrison EP,Kang HM,Korbel JO,Marchini JL,McCarthy S,McVean GA,Abecasis GR.2015.A global reference for human genetic variation.Nature,526(7571):68–74.
Abuzayed MA,Goktay M,Allmer J,Doganlar S,Frary A.2017.Development of genomic simple sequence repeat markers in faba bean by next-generation sequencing.Plant Molecular Biology Reporter,35(1):61–71.
Benjamini Y,Speed TP.2012.Summarizing and correcting the GC content bias in high-throughput sequencing.Nucleic Acids Research,40(10):e72.
Castagnone-Sereno P,Danchin EGJ,Deleury E,Guillemaud T,Malausa T,Abad P.2010.Genome-wide survey and analysis of microsatellites in nematodes,with a focus on the plant-parasitic speciesMeloidogyne incognita.BMC Genomics,11:598.
Chistiakov DA,Hellemans B,Volckaert FAM.2006.Microsatellites and their genomic distribution,evolution,function and applications:a review with special reference to fish genetics.Aquaculture,255(1–4):1–29.
Du LM,Li YZ,Zhang XY,Yue BS.2013.MSDB:a user-friendly program for reporting distribution and building databases of microsatellites from genome sequences.Journal of Heredity,104(1):154–157.
Engelhardt A,Muniz L,Perwitasari-Farajallah D,Widdig A.2017.Highly polymorphic microsatellite markers for the assessment of male reproductive skew and genetic variation in critically endangered crested macaques(Macaca nigra).International Journal of Primatology,38(4):672–691.
Fan ZX,Zhao G,Li P,Osada N,Xing JC,Yi Y,Du LM,Silva P,Wang HX,Sakate R,Zhang XY,Xu HL,Yue BS,Li J.2014.Whole-genome sequencing of Tibetan macaque(Macaca thibetana)provides new insight into the macaque evolutionary history.Molecular Biology and Evolution,31(6):1475–1489.
Gil J,Um Y,Kim S,Kim OT,Koo SC,Reddy CS,Kim SC,Hong CP,Park SG,Kim HB,Lee DH,Jeong BH,Chung JW,Lee Y.2017.Development of genome-wide SSR markers fromAngelica gigasnakai using next generation sequencing.Genes,8(10):238.
Guichoux E,Lagache L,Wagner S,Chaumeil P,Léger P,Lepais O,Lepoittevin C,Malausa T,Revardel E,Salin F,Petit RJ.2011.Current trends in microsatellite genotyping.Molecular Ecology Resources,11(4):591–611.
Gymrek M,Golan D,Rosset S,Erlich Y.2012.lobSTR:a short tandem repeat profiler for personal genomes.Genome Research,22(6):1154–1162.
Gymrek M,Willems T,Guilmatre A,Zeng HY,Markus B,Georgiev S,Daly MJ,Price AL,Pritchard JK,Sharp AJ,Erlich Y.2016.Abundant contribution of short tandem repeats to gene expression variation in humans.Nature Genetics,48(1):22–29.
Hall BG.2013.Building phylogenetic trees from molecular data with MEGA.Molecular Biology and Evolution,30(5):1229–1235.
International Human Genome Sequencing Consortium.2001.Initial sequencing and analysis of the human genome.Nature,409(6822):860–921.Ji HC,Li X,Yue SQ,Li JJ,Chen H,Zhang ZC,Ma B,Wang J,Pu M,Zhou L,Feng C,Wang DS,Duan JL,Pan DK,Tao KS,Dou KF.2015.Pig BMSCs transfected with human TFPI combat species incompatibility and regulate the human TF pathwayin vitroand in a rodent model.Cellular Physiology and Biochemistry,36(1):233–249.
Jia XD,Yang BD,Yue BS,Yin HL,Wang HX,Zhang XY.2011.Isolation and characterization of twenty-one polymorphic microsatellite loci in the Tibetan macaque(Macaca thibetana).Russian Journal of Genetics,47(7):884–887.Jia XD,Zhang XY,Wang HX,Wang ZK,Yin YH,Yue BS.2012.Construction of microsatellite-enriched library and isolation of microsatellite markers inMacaca thibetana.Sichuan Journal of Zoology,31(1):39–44.(in Chinese)
Jiang XL,Wang YX,Wang QS.1996.Taxonomy and distribution of tibetan macaque(Macacathibetana).Zoological Research,17(4):361–369.(inChinese)Jiang ZG,Jiang JP,Wang YZ,Zhang E,Zhang YY,Li LL,Xie F,Cai B,Cao L,Zheng G M,Dong L,Zhang ZW,Ding P,Luo ZH,Ding CQ,Ma ZJ,Tang SH,Cao WX,Li CW,Hu HJ,Ma Y,Wu Y,Wang YX,Zhou KY,Liu SY,Chen YY,Li JT,Feng ZJ,Wang Y,Wang B,Li C,Song XL,Cai L,Zang CX,Zeng Y,Meng ZB,Fang HX,Ping XG.2016.Red list of China’s vertebrates.Biodiversity Science,24(5):500–551.(in Chinese)
La Spada AR,Wilson EM,Lubahn DB,Harding AE,Fischbeck KH.1991.Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy.Nature,352(6330):77–79.
Levinson G,Gutman GA.1987.Slipped-strand mispairing: a major mechanism for dna sequence evolution.Molecular Biology and Evolution,4(3):203–221.
Li DM,Fan LQ,Ran JH,Yin HL,Wang HX,Wu SB,Yue BS.2008.Genetic diversity analysis ofMacaca thibetanabased on mitochondrial DNA control region sequences.DNA Sequence,19(5):446–452.
Li P,Yang CZ,Zhang XY,Li J,Yi Y,Yue BS.2014.Development of eighteen tetranucleotide microsatellite markers in Tibetan macaque(Macaca thibetana)and genetic diversity analysis of captive population.Biochemical Systematics and Ecology,57:293–296.
Li YC,Korol AB,Fahima T,Nevo E.2004.Microsatellites within genes:structure,function,and evolution.Molecular Biology and Evolution,21(6):991–1007.
Liu CC,Liu Y,Zhang XY,Xu XW,Zhao SH.2017a.Characterization of porcine simple sequence repeat variation on a population scale with genome resequencing data.Scientific Reports,7(1):2376.
Liu G,Zeng T,Yu WH,Yan NH,Wang HX,Cai SP,Pang IH,Liu XY.2011.Characterization of intraocular pressure responses of the Tibetan monkey(Macaca thibetana).Molecular Vision,17:1405–1413.
Liu SX,Hou W,Sun TL,Xu YT,Li P,Yue BS,Fan ZX,Li J.2017b.Genome-wide mining and comparative analysis of microsatellites in three macaque species.Molecular Genetics and Genomics,292(3):537–550.
MacDonald ME,Ambrose CM,Duyao MP,Myers RH,Lin C,Srinidhi L,Barnes G,Taylor SA,James M,Groot N,MacFarlane H,Jenkins B,Anderson MA,Wexler NS,Gusell JF,Bates GP,Baxendale S,Hummerich H,Kirby S,North M,Youngman S,Mott R,Zehetner G,Sedlacek Z,Poustka A,Frischauf AM,Lehrach H,Buckler AJ,Church D,Doucette-Stamm L,O’Donovan MC,Riba-Ramirez L,Shah M,Stanton VP,Stanton SA,Draths KM,Wales JL,Dervan P,Housman DE,Altherr M,Shiang R,Thompson L,Fielder T,Wasmuth JJ,Tagle D,Valdes J,Elmer L,Allard M,Castilla L,Swaroop M,Blanchard K,Collins FS,Snell R,Holloway T,Gillespie K,Datson N,Shaw D,Harper PS.1993.A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes.Cell,72(6):971–983.
Morin PA,Chambers KE,Boesch C,Vigilant L.2001.Quantitative polymerase chain reaction analysis of DNA from noninvasive samples for accurate microsatellite genotyping of wild chimpanzees(Pan troglodytes verus).Molecular Ecology,10(7):1835–1844.
Oliveira EJ,Pádua JG,Zucchi MI,Vencovsky R,Vieira MLC.2006.Origin,evolution and genome distribution of microsatellites.Genetics and Molecular Biology,29(2):294–307.
Osada N,Hettiarachchi N,Adeyemi Babarinde I,Saitou N,Blancher A.2015.Whole-genome sequencing of six Mauritian cynomolgus macaques(Macaca fascicularis)reveals a genome-wide pattern of polymorphisms under extreme population bottleneck.Genome Biology and Evolution,7(3):821–830.
Patel RK,Jain M.2012.NGS QC Toolkit:a toolkit for quality control of next generation sequencing data.PLoS One,7(2):e30619.
Powell W,Machray GC,Provan J.1996.Polymorphism revealed by simple sequence repeats.Trends in Plant Science,1(7):215–222.
Qi WH,Jiang XM,Du LM,Xiao GS,Hu TZ,Yue BS,Quan QM.2015.Genome-Wide survey and analysis of microsatellite sequences in bovid species.PLoS One,10(7):e0133667.
Qi WH,Yan CC,Li WJ,Jiang XM,Li GZ,Zhang XY,Hu TZ,Li J,Yue BS.2016.Distinct patterns of simple sequence repeats and GC distribution in intragenic and intergenic regions of primate genomes.Aging,8(11):2635–2654.
Qin YJ,Buahom N,Krosch MN,Du Y,Wu Y,Malacrida AR,Deng YL,Liu JQ,Jiang XL,Li ZH.2016.Genetic diversity and population structure inBactrocera correcta(Diptera:Tephritidae)inferred from mtDNAcox1and microsatellite markers.Scientific Reports,6:38476.
Qu JT,Liu J.2013.A genome-wide analysis of simple sequence repeats in maize and the development of polymorphism markers from next-generation sequence data.BMC Research Notes,6:403.
Rubinsztein DC,Amos W,Leggo J,Goodburn S,Jain S,Li SH,Margolis RL,Ross CA,Ferguson-Smith MA.1995.Microsatellite evolution—evidence for directionality and variation in rate between species.Nature Genetics,10(3):337–343.
Schl?tterer C,Tautz D.1992.Slippage synthesis of simple sequence DNA.Nucleic Acids Research,20(2):211–215.
Schl?tterer C.1998.Genome evolution:are microsatellites really simple sequences?Current Biology,8(4):R132–R134.
Schl?ttererC.2000.Evolutionary dynamics of microsatellite DNA.Chromosoma,109(6):365–371.
Sharma PC,Grover A,Kahl G.2007.Mining microsatellites in eukaryotic genomes.Trends in Biotechnology,25(11):490–498.
Simpson JT,Wong K,Jackman SD,Schein JE,Jones SJM,Birol I.2009.ABySS:a parallel assembler for short read sequence data.Genome Research,19(6):1117–1123.
Subramanian S,Mishra RK,Singh L.2003.Genome-wide analysis of microsatellite repeats in humans:their abundance and density in specific genomic regions.Genome Biology,4(2):R13.
Sunnucks P.2000.Efficient genetic markers for population biology.Trends in Ecology&Evolution15(5):199–203.
Taberlet P,Waits LP,Luikart G.1999.Noninvasive genetic sampling:look before you leap.Trends in Ecology&Evolution,14(8):323–327.
Team RC.2000.R Language Definition.Vienna,Austria:R Foundation for Statistical Computing.
Webster MT,Smith NGC,Ellegren H.2002.Microsatellite evolution inferred from human-chimpanzee genomic sequence alignments.Proceedings of the National Academy of Sciences of the United States of America,99(13):8748–8753.
Willems T,Gymrek M,Highnam G,The 1000 Genomes Project Consortium,Mittelman D,Erlich Y.2014.The landscape of human STR variation.Genome Research,24(11):1894–1904.
Xu LY,Haasl RJ,Sun JJ,Zhou Y,Bickhart DM,Li JY,Song JZ,Sonstegard TS,Van Tassell CP,Lewin HA,Liu GE.2016.Systematic profiling of short tandem repeats in the cattle genome.Genome Biology and Evolution,9(1):20–31.
Xue C,Raveendran M,Harris RA,Fawcett GL,Liu XM,White S,Dahdouli M,Rio Deiros D,Below JE,Salerno W,Cox L,Fan GP,Ferguson B,Horvath J,Johnson Z,Kanthaswamy S,Kubisch HM,Liu DH,Platt M,Smith DG,Sun BH,Vallender EJ,Wang F,Wiseman RW,Chen R,Muzny DM,Gibbs RA,Yu FL,Rogers J.2016.The population genomics of rhesus macaques(Macaca mulatta)based on whole-genome sequences.Genome Research,26(12):1651–1662.
Yang N,Zhou L,Liu XS,Zeng DW.2017.Isolation and characterization of novel Tibetan macaque(Macaca thibetana)microsatellite loci:cross-species amplification and population genetic applications.Biomedical Research,28(1):268–272.
Yi Y,Zeng T,Zhou L,Cai SP,Yin Y,Wang Y,Cao X,Xu YZ,Wang HX,Liu XY.2012.Experimental Tibetan monkey domestication and its application for intraocular pressure measurement.International Journal of Ophthalmology,5(3):277–280.
Zhong LJ,Zhang MW,Yao YF,Ni QY,Mu J,Li CQ,Xu HL.2013.Genetic diversity of two Tibetan macaque(Macaca thibetana)populations from Guizhou and Yunnan in China based on mitochondrial DNA D-loop sequences.Genes&Genomics,35(2):205–214.
Zhong XM,Peng JG,Shen QS,Chen JY,Gao H,Luan XK,Yan S,Y Huang X,Zhang SJ,Xu LY,Zhang XQ,Tan BCM,Li CY.2016.RhesusBase PopGateway:Genome-Wide population genetics atlas in rhesus macaque.Molecular Biology and Evolution,33(5):1370–1375.
Zimin AV,Cornish AS,Maudhoo MD,Gibbs RM,Zhang XF,Pandey S,Meehan DT,Wip fler K,Bosinger SE,Johnson ZP,Tharp GK,Mar?ais G,Roberts M,Ferguson B,Fox HS,Treangen T,Salzberg SL,Yorke JA,Norgren RB Jr.2014.A new rhesus macaque assembly and annotation for next-generation sequencing analyses.Biology Direct,9(1):20.

- Zoological Research的其它文章
- Variations in diet composition of sympatric Trachypithecus francoisi and Macaca assamensis in the limestone habitats of Nonggang,China
- Playing it cool:Characterizing social play,bout termination,and candidate play signals of juvenile and infant Tibetan macaques(Macaca thibetana)
- Female choice impacts resident male takeover in golden snub-nosed monkeys(Rhinopithecus roxellana)
- Ecology and social system of northern gibbons living in cold seasonal forests
- Extant primates and development of primatology in China:Publications,student training,and funding
- A road for a promising future for China’s primates:The potential for restoration