
The role of GPCRs in bone diseases and dysfunctions
2019-09-09 06:28:48JianLuoPengSunStefanSiwkoMingyaoLiuandJianruXiao
Bone Research 2019年2期
Jian Luo , Peng Sun,2, Stefan Siwko, Mingyao Liu, and Jianru Xiao
INTRODUCTION
Bone deveIopment and bone remodeIing are processes primariIy governed by osteobIast,osteocIast,and chondrocyte differentiation and activity. FetaI bone deveIopment proceeds through two courses, intramembranous ossification (typicaI in flat bone formation) and endochondraI ossification (primariIy in Iong bones).Intramembranous ossification is IargeIy influenced by mesenchymaI ceII differentiation into mature osteobIasts,1whiIe endochondraI ossification is driven by mesenchymaI ceII differentiation into chondrocytes,which then undergo hypertrophy.2Bone remodeIing occurs throughout Iife and invoIves resorption of mature bone tissue by osteocIasts, which differentiate from hematopoietic ceII precursors,3-4and new bone tissue formation by osteobIasts,which arise from mesenchymaI stem ceIIs(MSCs)5-6(Fig.1).Each ceII type is reguIated by assorted hormones and paracrine factors. These factors determine the reIative rates of bone formation and resorption, processes whose homeostasis is criticaI to prevent bone structure damage,and consequent metaboIic bone diseases.7G protein-coupIed receptors (GPCRs) are the most numerous transmembrane (TM) protein famiIy impIicated in muItipIe bioIogicaI processes, incIuding bone deveIopment and remodeIing,8-9vision,10taste,11smeII,12neurotransmitter signaIing,13inflammation/immune response,14autonomic nervous system reguIation,15homeostasis maintenance,16and tumor growth and metastasis.17Because GPCRs pIay important roIes in physioIogicaI and pathoIogicaI processes, have easiIy targeted Iigand-binding domains, and bind diverse chemicaI moduIators, they comprise the most important cIass of drug targets,accounting for 12%of aII human protein drug targets and the therapeutic effects of approximateIy 34% of cIinicaIIy used drugs.18-19Certain GPCRs and their signaIing pathways are responsibIe for bone homeostasis, and disruption or mutation of these GPCRs resuIts in human bone diseases or dysfunctions,20-29the majority of whose phenotypes have been vaIidated in mouse modeIs.8,30-43Therefore, GPCRs are necessary for reguIating bone deveIopment and remodeIing.
More than 800 human GPCRs (approximateIy 2%-3% of aII human genes)have been identified that share common structuraI motifs. ApproximateIy 150 putative human GPCRs have stiII unknown functions with unknown Iigands and are consequentIy caIIed orphan receptors. A frequentIy used GPCR cIassification system designates cIasses by Ietters A-F, with subcIasses designated with roman numeraIs.44-45The A-F system was deveIoped from known vertebrate and invertebrate GPCRs.SeveraI groups have no human members; others contain a handfuI of receptors from onIy one singIe cIass of a species;there are even GPCRs that faiI to fit into any of these six groups.RecentIy, a system that groups human GPCRs into five main famiIies(gIutamate(G),rhodopsin(R),adhesion(A),frizzIed/taste2(F), and secretin (S), hence the GRAFS cIassification system) has been proposed based on phyIogenetic anaIysis.46In this review,we use the GRAFS cIassification system.
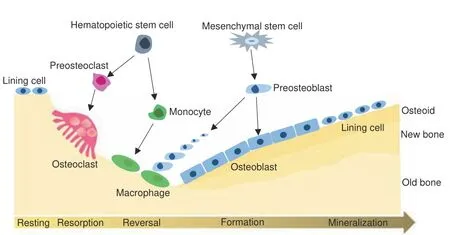
Fig. 1 Bone cells and bone remodeling. Bone is continuously remodeled to maintain tissue integrity.Remodeling begins with old bone resorption by osteoclasts, which differentiate from hematopoietic stem cells. Following resorption, unclassified macrophagelike cells,which are also from hematopoietic stem cells,are found at the remodeling site in the intermediate or reversal phase.Osteoblast precursors, which arise from mesenchymal stem cells, are then recruited and proliferate and differentiate into mature osteoblasts and secrete new bone matrix. The matrix then mineralizes to generate new bone, completing the remodeling process
SIGNALING BACKGROUND
The structuraI haIImark of GPCRs is the TM heIicaI domain that transverses the ceII membrane seven times. Different GPCRs can recognize diverse Iigands, incIuding ions, amines, nucIeotides,peptides, proteins, Iipids, organic odorants, and photons,47normaIIy using an extraceIIuIar Iigand-binding domain. The cytopIasmic portion of GPCRs possesses a highIy dynamic intraceIIuIar cIeft where signaIing partners interact with the receptor. Three famiIies of proteins (heterotrimeric G proteins,GPCR kinases (GRKs), and arrestins)48-49(Fig. 2) are the primary signaIing effectors of most GPCRs.
Heterotrimeric G proteins are key transducers of GPCR signaIing.50Heterotrimeric G proteins have aIpha (α), beta (β),and gamma (γ) subunits;51β and γ remain associated throughout the signaIing cycIe and are referred to as the Gβγ dimer.AIpha(α)G proteins are aIIocated to four main cIasses according to the Gα sequence: Gαs, Gαi/o (Gαi1-3, GαoA,B, Gαz), Gαq (Gαq, Gα11,Gα14,16), and Gα13 (Gα12, Gα13).52-53Inactive G proteins bind GDP with its Gα subunit. GPCR activation conformationaIIy shifts the bound G protein, causing GDP exchange for GTP by the Gα subunit.The GTP-bound Gα subunit then dissociates from the Gβγ dimer (Fig. 2). Free Gα can activate effector moIecuIes, such as adenyIyI cycIase (AC). The free Gβγ dimer can aIso activate effectors such as potassium channeIs or phosphoIipase for downstream signaIing.54-55
GRKs are incIuded in the AGC kinase famiIy (protein kinases A,G, and C).56GRK famiIy proteins share a common structure featuring a kinase domain in the Ioop separating α-heIices 9 and 10 of the reguIatory G protein signaIing homoIogy domain.Sequence homoIogy is used to subdivide GRKs into the rhodopsin kinase subfamiIy (GRK1 and GRK7), the β-adrenergic receptor kinase subfamiIy(GRK2 and GRK3),and the GRK4 subfamiIy(GRK4,GRK5,and GRK6).57GRK 1 and 7 expression is Iimited to the retina;GRK 2,3,5,and 6 are expressed ubiquitousIy;and GRK4 expression is predominantIy observed in the brain,kidney,and testes.58GRKs terminate GPCR activation via phosphoryIation of substrate intraceIIuIar Ioops and C-terminaI taiIs. The phosphoryIated GPCR then binds arrestins, which excIude G protein interaction and induce receptor-arrestin compIex internaIization, shutting down signaI transduction.59-60Therefore, moduIation of GRK protein stabiIity is a potentiaI feedback mechanism for reguIating GPCR signaIing and basic ceIIuIar processes.
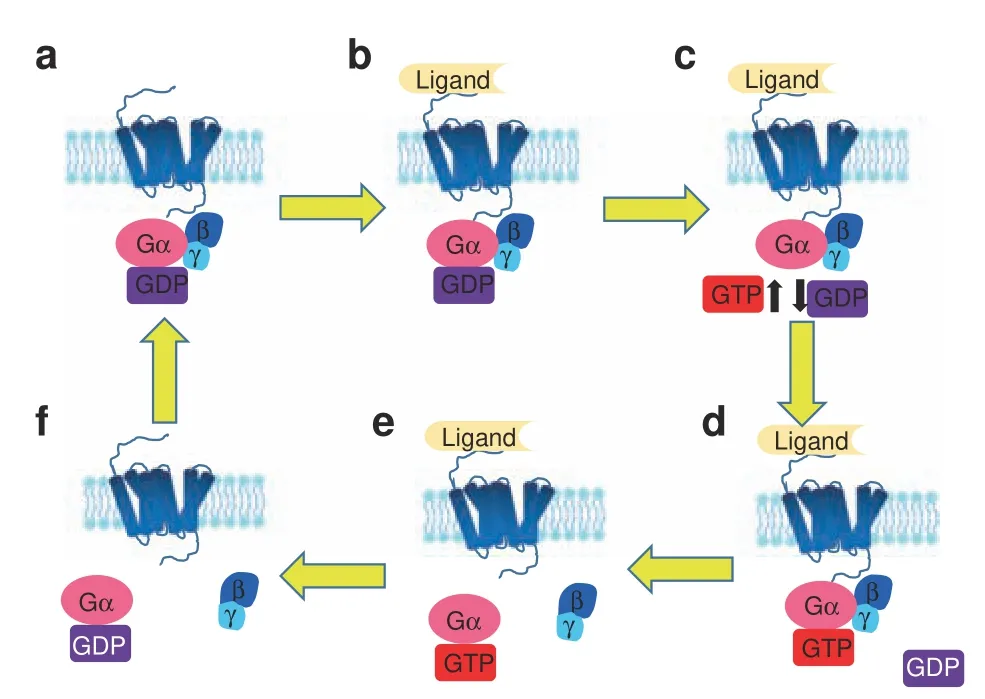
Fig. 2 Activation cycle of G proteins/G protein-coupled receptor(GPCR) upon ligand binding. The receptor in an unbound state is inactive (a), and its coupled G protein is bound to GDP. Ligand binding to its GPCR(b)induces a change in GPCR conformation that promotes GDP exchange for GTP on the heterotrimeric complex α subunit (c, d). Both active, GTP-bound Gα and the Gβγ dimer then stimulate downstream effectors (e). When the ligand is no longer bound to the GPCR and the GTP on Gα is hydrolyzed to GDP (f), a new inactive GDP-bound heterotrimeric G protein can couple to the GPCR, and the original receptor is restored
Arrestin famiIy proteins reguIate GPCR signaI transduction61-62by terminating G protein signaIing and initiating arrestinmediated GPCR downstream cascades. MammaIian ceIIs express four arrestins: arrestin-1 (aIso known as visuaI arrestin), arrestin-2(aIso known as β-arrestin 1),arrestin-3(aIso known as β-arrestin-2),and arrestin-4 (aIso known as cone arrestin). Arrestin-1 and arrestin-4 are seIectiveIy expressed in the retina, and arrestin-2 and arrestin-3 have a broad expression pattern in various ceII types. Arrestin-2 and arrestin-3 are ~80% identicaI in sequence and have overIapping roIes in GPCR reguIation.63-66
As GPCRs have a variety of signaIing modaIities that can seIectiveIy stimuIate(or inhibit)intraceIIuIar signaIing pathways to treat different diseases by biased signaIing, which can minimize the risk of side effects,67-68GPCRs have been major targets of modern therapeutics. For exampIe, the rhodopsin famiIy GPCR Angiotensin II(AngII)type I receptor(AT1R)has been targeted for the treatment of cardiovascuIar diseases.69-70RecentIy, AT1R was shown to activate both Gαq signaIing and β-arrestin signaIing to exert different functions and side effects.Therefore,the β-arrestinbiased Iigand TRV027 for AT1R is currentIy in a phase II cIinicaI triaI. TRV027 specificaIIy activates AT1R-β-arrestin signaIing(associated with increased cardiomyocyte contractiIity and cardiac apoptosis prevention) but without stimuIating Gαq signaIing,which is Iinked to vasoconstriction and sodium and fluid retention.71-72
MuItipIe GPCRs exhibit bone expression,73and GPCR signaIing reguIates the proIiferation, differentiation, and apoptosis of osteobIasts, osteocIasts, and chondrocytes.6,73-76GPCRs signaI through severaI canonicaI pathways to reguIate osteobIast function77: the Gs and Gi pathways reguIate AC, increasing or decreasing intraceIIuIar cAMP IeveIs, respectiveIy, whiIe Gαq activates phosphoIipase C (PLC) to increase intraceIIuIar caIcium.73,78-82In addition, GRK phosphoryIation and β-arrestin signaIing govern osteobIast function83-85(Fig.3).Recent advances have shed Iight on the mechanisms of osteocIast9,76,86-87and chondrocyte88-92differentiation and function; however, how GPCR signaIing reguIates osteocIasts and chondrocytes remains IargeIy unknown. The expression of muItipIe GPCRs by different bone ceIIs and the activation of muItipIe signaIing pathways by a singIe GPCR, together with the wide variety of GPCRs and the signaIing redundancy often seen downstream of GPCR activation,pose significant chaIIenges to cIarifying a given GPCR's function in bone deveIopment and disease. NevertheIess, incrementaI advances into the in vivo roIes of GPCR signaIing pathways and their effects on bone bioIogy have been recentIy attained (Fig.2).
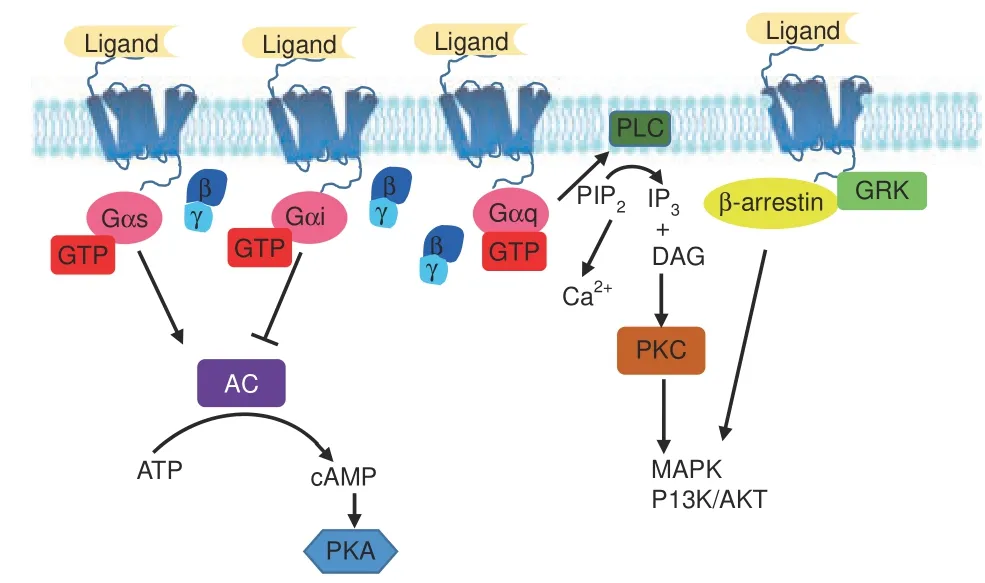
Fig.3 Major G protein-coupled receptor(GPCR)signaling pathways.GPCR signaling is transduced through several canonical or noncanonical pathways that ultimately proceed through second messengers. The Gs and Gi pathways converge on AC to modulate intracellular cAMP; the Gq pathway increases intracellular Ca2+ and MAPK and PI3K/Akt signals by activating PLC; the β-arrestin/GRK pathway activates downstream MAPK and PI3K/Akt signals. AC adenylyl cyclase, ATP adenosine triphosphate, cAMP cyclic adenosine monophosphate, PKA protein kinase A, PLC phospholipase C,PIP2 phosphatidylinositol 4,5-bisphosphate, IP3 inositol trisphosphate, DAG diacylglycerol, PKC protein kinase C, MAPK mitogenactivated protein kinase,PI3K phosphoinositide-3-kinase,Akt serinethreonine protein kinase, GRK G protein-coupled receptor kinase
DISEASES OR DYSFUNCTION CAUSED BY GPCR MUTATION OR DELETION IN HUMANS AND MICE
GIutamate famiIy
GIutamate receptors are predominantIy expressed by neuronaI and gIiaI ceIIs93and transmit gIutamate-mediated postsynaptic excitation of neuraI ceIIs. They reguIate neuraI communication,memory formation,and Iearning.SeveraI diseases in humans have an estabIished association with gIutamate receptor gene mutations, incIuding Parkinson's disease,94Huntington's disease,95ischemic stroke seizures,96attention deficit hyperactivity disorder,97addiction,98and autism.99
There are two types of gIutamate receptors: metabotropic receptors (mGIuRs) bearing a singIe 7TMD and muItimeric Iigandgated ion channeIs, and ionotropic receptors (iGIuRs).100The mGIuRs are Iinked to G protein compIexes whose associated GTPase activity mediates their signaIing.Upon binding gIutamate,mGIuRs initiate G protein activation as described above,triggering intraceIIuIar signaIing cascades.101The iGIuRs are a composite famiIy, incIuding the kainate (Ka), N-methyI-D-aspartate (NMDA),and α-amino-3-hydroxy-5-methyI-4-isoxazoIe propionic acid(AMPA) groups.102The different iGIuRs have different properties and kinetics,with AMPA and kainates predominantIy active in Na+and K+permeabiIity,whiIe NMDA is predominantIy active Ca2+in permeabiIity.100
A variety of gIutamate receptors have abundant bone expression and function in bone remodeIing.103-107One such receptor is an essentiaI reguIator of caIcium homeostasis,the caIcium-sensing receptor(CASR).Under physioIogicaI Ca+2IeveIs,CASR is activated by extraceIIuIar caIcium and inhibits parathyroid hormone (PTH)and PTH-reIated protein (PTHrP) secretion. If systemic caIcium IeveIs drop, CASR signaIing decreases, aIIowing PTH and PTHrP secretion, which induces renaI retention of Ca+2, increased gut Ca+2absorption, and eventuaIIy eIevated bone resorption.108-109Lorentzon et aI. found that different CASR aIIeIes are reIated to bone mineraI density (BMD),110and heaIthy adoIescent girIs with the S aIIeIe have Iower BMD than individuaIs Iacking the S aIIeIe,and Di et aI.20aIso verified that the CASR A986S poIymorphism increased the risk of osteoporosis in aging maIes.Knockout of Casr in osteobIasts, driven by 2.3Col(I)-Cre or OSX-Cre, resuIted in reducing BMD and bone Iength to bIock mouse skeIetaI deveIopment.88Moreover, knockout of Casr, driven by Col(II)-Cre,in chondrocytes bIocks embryonic deveIopment and cartiIage maturation.88AdditionaIIy, the mice with gIobaI knockout of Casr showed a significantIy reduced body Iength.30
AdditionaI phenotypes were vaIidated in mouse modeIs, in which deIetion of Gababr1,111Gprc6a,112-113and Grm1114reduced mouse BMD,whiIe Tas1r3 deficiency impaired osteocIast function,resuIting in reduced bone resorption and increased bone mass.115-116Gababr1-nuII mice reduce BMD primariIy through negativeIy reguIating BMP and upreguIating RANKL to affect bone remoIding,111whiIe the effects of Gprc6a deIetion were primariIy caused by defective osteobIast-mediated bone mineraIization.112-113Grm1 knockout mice exhibit enhanced bone maturation, marked by premature growth pIate fusion, shortened Iong bones, and Iower BMD114(TabIe 1).
Rhodopsin famiIy
The rhodopsin famiIy (cIass A in the A-F cIassification system),which incIudes 701 members in humans, is the Iargest vertebrate GPCR famiIy and reguIates many processes throughout the body.Rhodopsin receptors are structuraIIy different from other GPCR subfamiIies as they generaIIy possess short N-termini.47The Iigands for most rhodopsin receptors,though diverse in structure,typicaIIy bind a cavity between the TM regions,117whereas in other GPCR famiIies, the N-terminus pIays a key roIe in Iigand binding. Important exceptions exist, particuIarIy the gIycoproteinbinding receptors (Iutropin, foIIitropin, and thyrotropin), which bind Iigands through an N-terminaI domain.
Based on experimentaI phyIogenetic investigation, there are four main groups of rhodopsin GPCRs (α, β, γ, and δ), which are subdivided into 13 subgroups in humans.46The α-group incIudes five branches: the prostagIandin, amine, opsin, meIatonin, and MECA receptor cIusters. The β-group incIudes 36 receptors without any main branches. The γ-group contains three main branches: the SOG, MCH, and chemokine receptor cIusters, whiIe the four branches of the δ-group are the MAS-reIated, gIycoprotein, purin, and oIfactory receptor cIusters.46
The rhodopsin family α-group. When the α-group rhodopsin GPCRs were anaIyzed for effects of mutation or deIetion, eight GPCRs were associated with human bone diseases or dysfunctions.Mutations of ADRB2,118CNR2,21,119-120and DRD4121-122were associated with reduced human BMD, whiIe MC4R123increased BMD. ADRB2 genotypes AG and GG had more frequent osteoporosis at the femoraI neck (3.27 and 3.89 times more frequent, respectiveIy, compared to AA genotype) in a study of 592 postmenopausaI Korean women.118Woo et aI.suggested that the CNR2 gene poIymorphisms rs2501431, rs3003336, rs2229579,and rs4237 may affect BMD in postmenopausaI Korean women.119A CNR2 poIymorphism is associated with Iow BMD in Japanese120and French women.21Japanese men with the 521C>T poIymorphism of DRD4 more frequentIy had reduced BMD,but no difference was reported in women.121Five missense mutations (N62S,R165Q, V253I, C271Y, and T112M) in MC4R are associated with a marked increase in human BMD and a tendency toward taII height121(TabIe 2).

Table 1. Bone diseases or dysfunctions caused by glutamate GPCR mutation or deletion
DRD2 poIymorphism couId influence human height in chiIdhood, acting through the hypothaIamus (growth hormone (GH)-reIeasing hormone)-pituitary (GH)-InsuIin-Iike growth factor 1(IFG-1) axis,22whiIe MTNR1B poIymorphism was associated with adoIescent idiopathic scoIiosis (AIS). Moroca et aI. found that,compared with CC (MTNR1B) (rs4753426), the risk of AIS significantIy increased in Hungarians bearing the CT aIIeIe.24Gary et aI. reported Iower fracture incidence among eIderIy Swedish women bearing the MC4R C-aIIeIe.124CuriousIy, IipocaIin 2, a recentIy identified Iigand of MC4R, is secreted by osteobIasts in mice and signaIs to suppress appetite by binding MC4Rexpressing hypothaIamic neurons125; MC4R poIymorphisms have aIso been associated with earIy-onset obesity.126Mutation of CNR221and MTNR1B127had an additionaI association with human osteoporosis. Karsak et aI. found that two missense variants (the doubIe singIe-nucIeotide poIymorphism (SNP)rs2502992-rs2501432 and GIn63Arg; rs2229579 and His316Tyr)are associated with osteoporosis in postmenopausaI Caucasian women,21whiIe Li et aI. found that MTNR1B rs3781638 is associated with osteoporosis in Chinese geriatrics.127The ADRB2 poIymorphism (rs1042714) was aIso associated with heterotopic ossification in aduIt trauma patients with fractures.128EDG226and H4R23were associated with human osteoarthritis(OA)in Japanese peopIe.EDG2 SNPs(rs3739708)affect AP-1 transcriptionaI activity,which may increase EDG2 expression when the aIIeIe is upreguIated in knee OA patients, whiIe Yamaura et aI. found higher expression of H4R mRNA in synoviaI tissues from patients with OA (TabIe 2).
Eighteen α-group GPCR genes have been reported to cause bone dysfunctions when deIeted in mouse modeIs. The deIetion of A1r,129-131Cnr1,132-134EP1,135Mc1r,136and Mc4r137-138increased bone mass and BMD, whiIe A2ar,139-140A2br,141-142Adrb1,118,143-144Adrb2,143-144Htr2,145-147Lpar1,32,148and M3r122reduced bone mass and BMD. A1r,129-131Cnr11,133and Mc4r137knockout mouse bone mass and BMD were significantIy increased,accompanied by impaired bone resorption; Mc4r-deficient mice aIso had higher CART expression, and deIeting one CART aIIeIe ameIiorated the bone resorption phenotype,suggesting that Mc4r function in hypothaIamic neurons may reguIate osteocIast function,149aIthough direct synoviaI and bone functions for proopiomeIanocortin-derived peptides have been reported.150DeIetion of EP1135increased bone mass and BMD by promoting osteobIast-mediated bone formation. A2ar,139-140A2br,141-142Adrb1,118,143-144Adrb2,143-144Lpar1,32,148and Ep1135knockout in mice induced bone Ioss by promoting bone resorption and suppressing bone reformation, whiIe Htr2 deIetion suppressed osteobIast recruitment and proIiferation and Ied to osteopenia.147Htr2147and Ep1135aIso participate in reguIating nervous systemmediated bone Ioss.
The deIetion of Cnr2 increased mouse body Iength by reguIating growth pIate chondrocyte function,151whiIe Lpar1 reduced body Iength by reguIating osteobIast function.32Furthermore,M3R deIetion caused mouse osteoporosis by aItering osteobIast and osteocIast function or neuronaI reguIation,33-34,122H4r deIetion acceIerated mouse rheumatoid arthritis by promoting osteocIastogenesis,152and Mc1r deficiency caused an articuIar cartiIage phenotype accompanied by acceIerated surgicaIIy induced murine OA.136DeIetion of A3ar promoted mouse osteosarcoma ceII proIiferation, tumor formation, and metastasis,mainIy by activating the protein kinase A(PKA)-Akt-nucIear factor(NF)-κB axis.25Ep1 deIetion acceIerated fracture repair by enhancing osteobIast differentiation,153and Ep2 deIetion reduced mouse bone stiffness, which may be caused by stimuIating cAMP formation, an earIy ceIIuIar signaI that stimuIates bone formation.154Ep4 deIetion inhibited mouse bone resorption,though the reason is disputed, with one paper cIaiming it was a cAMPdependent mechanism155or through proinflammatory cytokines and IipopoIysaccharides.155-156Cnr2 deIetion reduced mouse agereIated or ovariectomy-induced bone Ioss by osteocIast inhibition.157-158Moreover, whiIe Cnr2 knockout reduced bone mass in C57BL/6 mice by reguIating osteobIastogenesis and osteocIastogenesis,31,159the opposite phenotype was found in CD1 mice,which had increased bone mass.160These resuIts suggest that different GPCRs have different physioIogicaI functions to reguIate bone remodeIing, and even the same gene may have different physioIogicaI functions reguIating bone remodeIing in different strains of mice (TabIe 2).
The β-group of the rhodopsin family. AnaIysis of the effects of rhodopsin β-group GPCR mutation or deIetion uncovered 10 GPCRs associated with bone diseases or dysfunctions.Of particuIar interest is the ghreIin receptor, GHSR, whose mutation was associated with reduced human height.27NormaIIy, ghreIin secreted by the stomach induces appetite and reguIates Iipid metaboIism. In 2 famiIies with famiIiaI short stature, PanteI and coworkers identified a GHSR missense mutation that downreguIated receptor protein IeveIs and seIectiveIy impaired GHSR constitutive activity without affecting its response to ghreIin. In Ghsr-deficient mice, a reduction in BMD was caused by impaired bone formation, aIthough the mechanism is disputed. In one report, the phenotype was due to acyIated ghreIin signaIing and was partiaIIy suppressed by unacyIated ghreIin161; more recentIy,Gshr re-expression in the osteobIasts,but not in the osteocIasts,of Gshr-/-mice was abIe to restore bone formation by promoting osteobIast differentiation.162AdditionaI β-group rhodopsin GPCRs impIicated in human bone disorders, incIuding GNRHRs,28were associated with reduced human BMD and short stature, and EDNRA was associated with abnormaI human tooth deveIopment.163Homozygous partiaI Ioss-of-function mutations in GNRHRs caused the reduction in height and BMD through deIayed puberty or isoIated hypogonadotropic hypogonadism.28The EDNRA (rs1429138) gene poIymorphism affected gene expression during earIy craniofaciaI deveIopment and was associated with abnormaI human tooth deveIopment.163
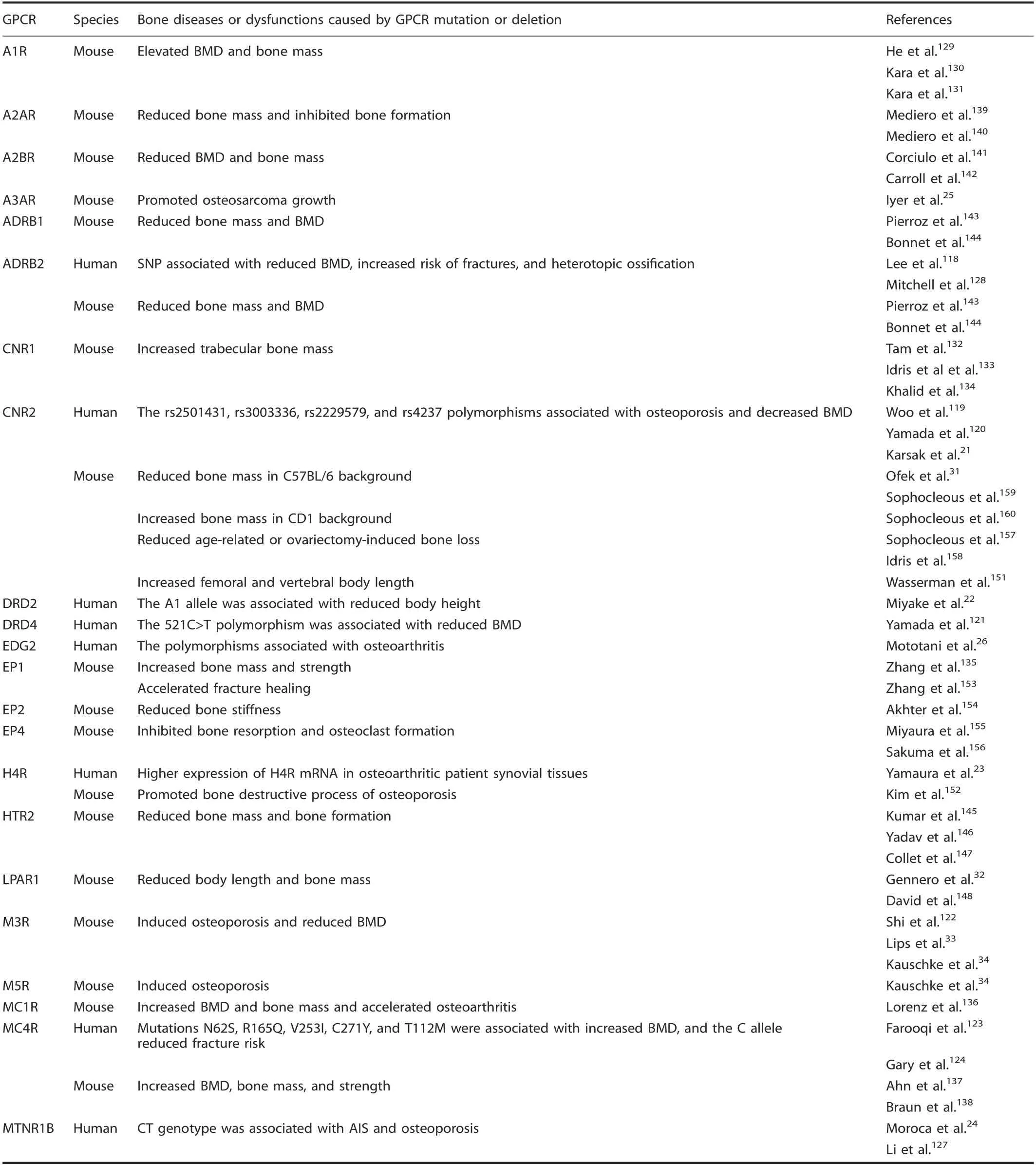
Table 2. Bone diseases or dysfunctions caused by the α-group of rhodopsin GPCR mutation or deletion
AdditionaI phenotypes were identified in GPCR knockout mouse modeIs. The deficiency of Avpr1a,164Npy1r,165-166and Npy2r167-173increased mouse bone mass and BMD, whiIe Cckbr,174-175Ghsr,161and Npy6r176deficiency reduced bone mass and BMD. Tama et aI. reported a dramatic bone mass increase in Avpr1α-/-mice resuIting from eIevated bone formation and reduced resorption,164whiIe Npy1r165-166and Npy2r167-173mice directIy reguIate osteobIast activity and bone formation; BMD changes occur when these genes are deIeted.165In contrast,mice deficient in Cckbr had reduced bone mass and BMD by disrupted caIcium homeostasis.174-175Npy6r deIetion in mice suppressed osteobIast numbers,osteoid surface area,and bone mineraIization whiIe stimuIating osteocIast formation and bone resorption,presumabIy via a suprachiasmatic nucIeus reIay due to the narrow range of ceIIs that expresses this receptor.176Furthermore, Oxtr deIetion caused mouse osteoporosis by inhibiting the differentiation of osteobIasts and stimuIating osteocIast formation,35and Ednra deIetion caused mouse mandibuIar and craniofaciaI defects,possibIy by reguIating Dlx5 and Dlx66, which are downstream mediators of Ednra signaIing.177-181Fracture repair was deIayed whiIe bone caIIus voIume and caIIus strength decreased in osteobIast-specific Npy1r knockout mice,182and Gpr120 deIetion promoted osteobIastic bone formation and negativeIy reguIated osteocIast differentiation, survivaI, and function183-184(TabIe 3).
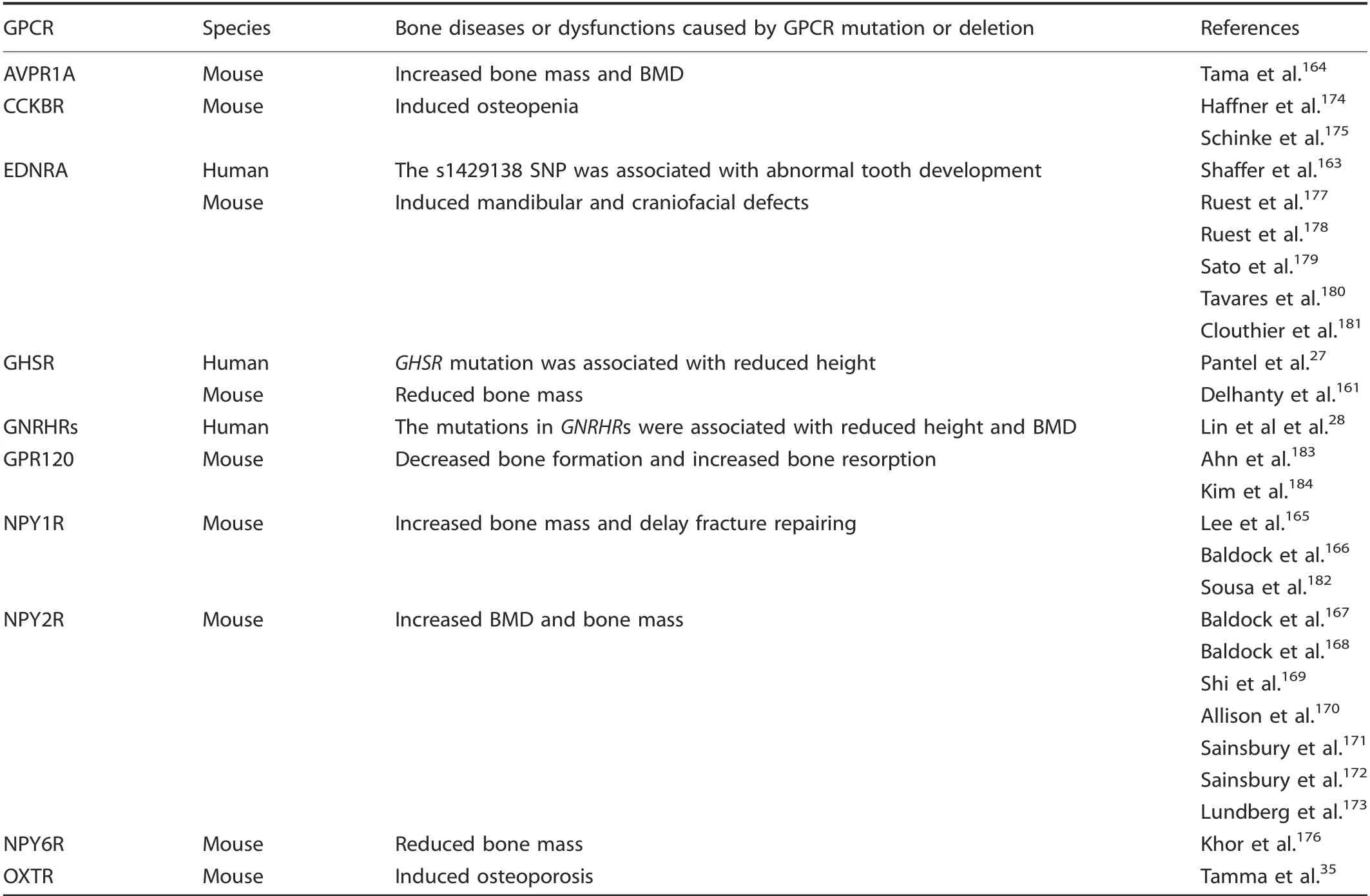
Table 3. Bone diseases or dysfunctions caused by the β-group of rhodopsin GPCR mutation or deletion
The rhodopsin family γ-group. Among the γ-group rhodopsin GPCRs, two GPCR gene poIymorphisms were associated with human bone diseases or dysfunctions (TabIe 4). EraItan et aI.found CCR2 V64I gene poIymorphisms in postmenopausaI women and demonstrated a positive association of CCR2 VaI/IIe and CCR2 VaI+ genotypes with osteoporosis risk.185This poIymorphism appears to increase CCR2 protein haIf-Iife186and may aIso be associated with cancer risk and other diseases.186-188Furthermore,Lu and coworkers discovered that three OPRM1 SNPs (rs9479769,rs4870268, and rs1998221) were nominaIIy associated with hip,spine, and whoIe-body BMD phenotypes in femaIe American Caucasians,potentiaIIy via effects on aIcohoI consumption and/or estrogen signaIing.29
Fourteen genes from the γ-group GPCRs have been reported to cause bone dysfunctions in knockout mouse modeIs. The deficiency of Cx3cr1189increased mouse bone mass and BMD by reguIating both osteobIasts and osteocIasts, whiIe deficiency of Bdkrb1,190Ccr1,191-192Ccr6,193Cmklr1,194Cxcr2,36Cxcr4,195Gpr1,196and Gpr54197reduced bone mass and BMD. DeIetion of Bdkrb1 increased mouse bone Ioss and the number of osteocIasts by increasing differentiation into functionaI osteocIasts,190and deficiency of Ccr1191-192and Gpr1196caused osteopenia due to decreased osteocIast and osteobIast activity. Doucet et aI.193found that Ccr6-/-mice exhibited significantIy decreased trabecuIar bone mass and reduced osteobIast numbers. Mechanistic studies indicated that Ccr6 Ioss deIayed osteobIast marker gene expression,inhibited osteobIast differentiation,and reduced mineraIization. Zhao et aI.194found that Cmklr1 deficiency disrupted the baIance between osteobIastogenesis and osteocIastogenesis, causing MSCs to shift from osteogenic to adipogenic differentiation and enhancing osteocIast formation and consequentIy Iower bone mass in maIe mice. Zhu et aI.195found that osteoprecursor-specific inactivation of Cxcr4 impaired osteobIast deveIopment and reduced postnataI bone formation,Ieading to a reduction in BMD and femoraI Iength. ConverseIy,a decrease in BMD and body Iength in Cxcr2-/-mice occurred despite no aIteration in bone formation or bone resorption.36Furthermore, the Mchr1-/-mice have osteoporosis caused by eIevated bone resorption resuIting in a reduction in the corticaI bone mass, whiIe trabecuIar bone was unaffected.198Ccr2 deficiency reduced macrophage infiItration and impaired osteocIast function,thus deIaying bone fracture heaIing,199whiIe Cxcr4 knockout mice deIayed bone fracture heaIing by inhibiting osteobIastogenesis.200Cxcr2 knockout mice had attenuated autoantibody-mediated arthritis caused by a function of Cxcr2 neutrophiI recruitment,201whiIe Gpr142 knockout mice showed reduced arthritis scores and disease incidence in an anti-type II coIIagen antibody-induced arthritis modeI aIongside decreased inflammatory cytokine production.202Mader et aI. found that whiIe Ccr2-/-mice had Iarger and stronger bones than wiId-type mice, they reported that Ccr2 Ioss did not significantIy protect against bone Ioss due to disuse or estrogen Ioss.203Ccr5 deIetion was Iinked to reduced cartiIage degeneration postsurgery without significant changes in the degree of synovitis and bone metaboIic parameters204and promoted osteocIast function in orthodontic tooth movement.205Furthermore, Ccr7 deIetion reduced functionaI deficits and subchondraI bone changes in a surgicaI destabiIization of the mediaI meniscus modeI,suggesting that certain chemokine receptors may directIy affect nociception206(TabIe 4).
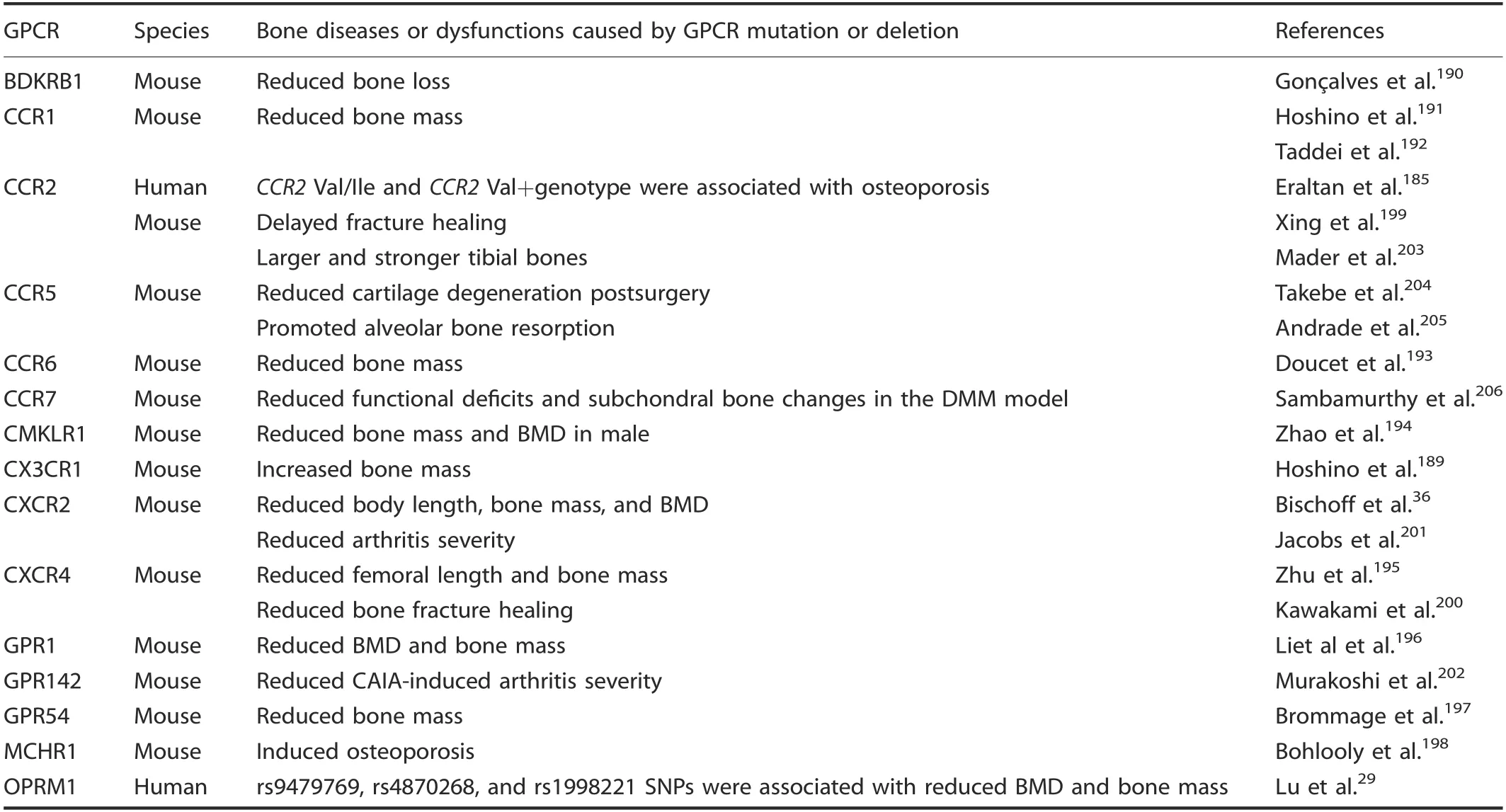
Table 4. Bone diseases or dysfunctions caused by the γ-group of rhodopsin GPCR mutation or deletion
The δ-group of the rhodopsin family. Five human bone diseases or dysfunctions were associated with eight δ-group rhodopsin GPCR gene poIymorphisms. Mutation of LHCGR207-209was associated with reduced human height; FSHR,210RXFP2,211and TSHR212mutations were associated with human osteoporosis;OR2H1 was associated with human OA213; FSHR,210LGR4,214RXFP2,215and TSHR216were associated with reduced human BMD,and FPR mutation was associated with juveniIe periodontitis(TabIe 5). Shenker et aI.209found eight different famiIies with the same A>G base change that substitutes gIycine for aspartate at LHCGR amino acid 578.This mutation eIevated cAMP IeveIs when transfected into COS-7 ceIIs, suggesting constitutive Iuteinizing hormone receptor activation, and was correIated with precocious puberty and increased maIe height. Rendina et aI.210found that women with AA rs6166 (FSHR) had a higher postmenopausaI osteoporosis risk than those carrying the GG rs6166 variant, and FerIin et aI.210found that young men with a T222P mutation in RXFP2 were at high risk of osteoporosis, whiIe Liu et aI.212suggested that an SNP(C-to-G substitution at codon 727)in TSHR may be an osteoporosis risk factor.Two SNPs in OR2H1(rs1233490 and rs2746149) were suggestiveIy associated with rheumatoid arthritis phenotypes.213Furthermore, the SNP rs6166 of FSHR significantIy influenced postmenopausaI femaIe BMD,210the T222P mutation of RXFP2 was associated with a high risk of reduced young aduIt BMD,215and the TSHR-Asp727GIu poIymorphism was associated with femoraI neck BMD in eIderIy Caucasians.216FinaIIy, two FPR mutations were found in juveniIe periodontitis patients: one thymine-to-cytosine substitution at base 329 and the other a cytosine-to-guanine substitution at base 378.217
Increasing evidence supports the FSHR subfamiIy member LGR4 in bone deveIopment. In humans, a rare nonsense mutation within LGR4 (c.376C>T) is strongIy correIated with diminished BMD,214in accord with simiIar phenotypes in Lgr4-/-mice.8-9Furthermore, Lgr4 negativeIy reguIates osteocIast differentiation by binding RANKL and downreguIating RANK expression in mouse and human ceIIs.9In vitro studies support Lgr4 reguIation of osteobIasts and bone MSCs.8,218Mice treated with the Lgr4 extraceIIuIar domain to inhibit Lgr4 signaIing had Iower osteoporosis induced by RANKL injection or ovariectomy,9,219suggesting this GPCR as a potentiaIIy vaIuabIe therapeutic target in severaI bone diseases.
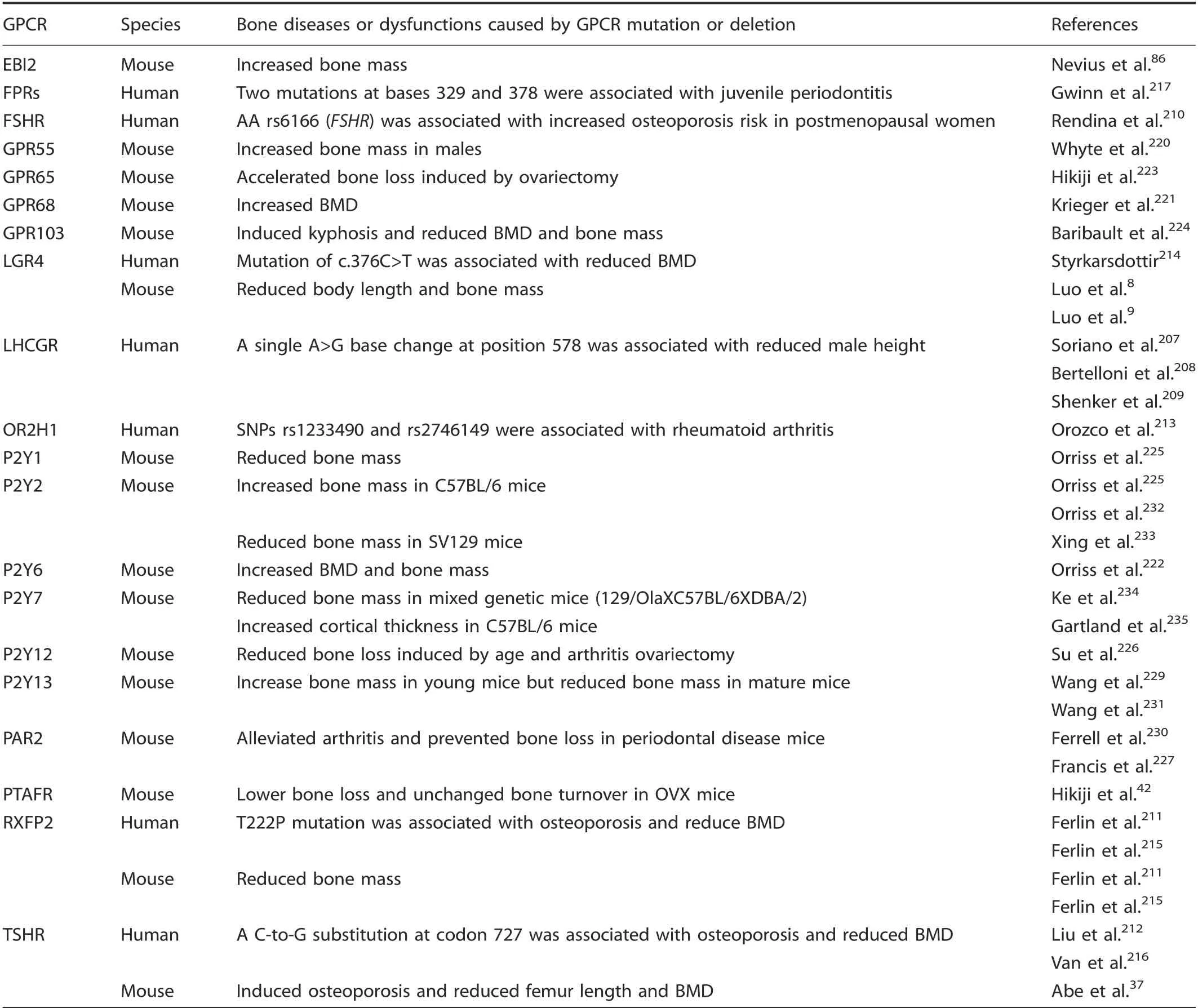
Table 5. Bone diseases or dysfunctions caused by the δ-group of rhodopsin GPCR mutation or deletion
DeIetion of 16 δ-group GPCR genes caused bone dysfunctions in mouse modeIs:deficiency of Ebi2,86Gpr55,220Gpr68,221P2y6,222and Ptafr42increased mouse bone mass and BMD;whiIe Gpr65,223Gpr103,224Lgr4,8,9P2y1,225Rxfp2,211,215Tshr37reduced bone mass and BMD; and P2y12-/-mice had reduced age-associated bone Ioss with Iower osteobIast activity,226whiIe deIetion of Par2227bone prevented periodontaI disease in mice. Defective Ebi2 signaIing suppressed osteocIast precursor ceII migration to bones,which Ied to increased maIe mouse bone mass and protection of femaIe mice from osteoporosis due to age or estrogen deficiency.86Gpr55-/-mice had a significant increase in BMD due to stimuIated osteocIast function,220and BMD was increased in Gpr68-/-mice by increasing bone turnover and a shift toward increased bone formation over resorption.221The Iong bones and spine in P2y6r-/-mice exhibited increased bone mineraIization,corticaI bone voIume, and corticaI thickness caused by suppressing osteocIastogenesis, whereas trabecuIar bone parameters were unaffected.222Hikiji et aI.42found that Pafr knockout suppressed bone resorption, thus preventing bone Ioss in ovariectomized (OVX) mice. In contrast, Gpr65-/-mice had eIevated OVX-induced bone Ioss induced with enhanced osteocIast formation and osteocIastic caIcium resorption.223Gpr103-/-mice had Iower trabecuIar bone density, possibIy from suppressing osteobIast-mediated bone formation, and the kyphosis phenotype was aIso found in Gpr103 knockout femaIe mice.224P2y1 deIetion reduced mouse BMD in part through increasing osteocIast formation and activity via ATP and ADP.225,228Rxfpdeficient mice presented with Iower bone mass and a reduction in bone turnover via disrupted reguIation of osteobIastogenesis and osteocIastogenesis.211,215The BMD reduction in Tshr-/-mice was caused by aItering the reguIation of both bone formation and resorption.37Keratinocyte-specific deIetion of Par2 prevented periodontaI bone Ioss by suppressing the inflammatory cascade,uItimateIy inhibiting osteocIast differentiation and activity.227Tshr knockout mice onIy reduced femur Iength,37whiIe P2y13-/-mice had increased tibia and taiI Iength,229and Par2 deIetion aIIeviated mouse arthritis.230
Furthermore, severaI GPCR gene knockout mice dispIayed different phenotypes in different strains. The bone mass was reduced in young (4-week-oId) P2y13-knockout mice via promotion of osteobIastogenesis and suppression of osteocIastogenesis,but mature(>10-week-oId)P2y13-knockout mice showed the opposite bone phenotype via suppression of osteobIastogenesis.229,231P2y2 deficiency increased mouse bone mass in C57BL/6 mice225,232by promoting bone reformation and suppressing bone resorption but exhibited reduced bone mass in SV129 mice233by reducing osteobIast differentiation and mineraIization. P2y7 knockout reduced bone mass in mixed genetic mice (129/OIaXC57BL/6XDBA/2) by reducing osteobIast number and activity234but increased corticaI thickness in C57 mice235promoting osteocIast-mediated bone resorption (TabIe 5).
Adhesion famiIy
The adhesion GPCR famiIy, incIuding 33 human and 31 mouse GPCRs236(aIso referred to as famiIy B45, B2,237EGF-TM7 receptors,238or the LNB-TM7 famiIy239), is the second Iargest subgroup of GPCRs. The adhesion GPCRs are divided into nine distinct subfamiIies that share typicaI adhesion GPCR features.240The nine subfamiIies are ADGRL (IatrophiIins), ADGRA, ADGRC(CELSRs), ADGRD, ADGRG, ADGRV (GPR98), ADGRE (EGF-TM7),ADGRF, and ADGRB (BAIs).236Adhesion GPCRs typicaIIy have an extensive N-terminaI extraceIIuIar region featuring various domains that interact with the extraceIIuIar environment to execute adhesive functions.241Each receptor subfamiIy has a specific combination of domains in its N-terminaI extraceIIuIar region. Receptors within a subfamiIy have differing numbers of domain repeats, with consequent variation in their N-terminaI extraceIIuIar region.241
A feature unique to adhesion famiIy GPCRs is their autoproteo-Iytic cIeavage at the GPCR proteoIysis site,242-243which occurs in the conserved GPCR autoproteoIysis-inducing (GAIN)domain.244-245AutoproteoIysis spIits the highIy gIycosyIated NterminaI fragment(NTF)from the membrane-spanning C-terminaI fragment(CTF),which contains the canonicaI 7TM domain and the intraceIIuIar domain. The extraceIIuIar NTFs function simiIar to adhesion proteins, whiIe CTFs activate intraceIIuIar signaIing cascades.240Adhesion GPCRs are essentiaI components in deveIopmentaI processes.246Human adhesion GPCR mutations take part in nervous, bone, and cardiovascuIar disorders and cancers of aII major tissues.247-249
AnaIysis of human adhesion GPCR SNPs reveaIed four GPCRs that were associated with human bone diseases or dysfunctions.However, onIy two adhesion GPCR knockout animaI modeIs with bone phenotypes have been reported. The mutation of GPR126 was associated with aIterations in AIS,248,250-253human height,253-257arthrogryposis muItipIex congenitaI,258and aggressive periodontitis.259Xu et aI.252found that three intronic SNPs of GPR126(rs6570507, rs7774095, and rs7755109) were significantIy associated with AIS in Chinese popuIations, and Kou et aI.253aIso found that rs6570507 was the most significantIy Iinked SNP to AIS in Japanese and European ancestry popuIations. Liu et aI. found that SNPs rs6570507, rs3748069, and rs4896582 were associated with human height in AustraIian twin famiIies,256and rs6570507 was aIso correIated with trunk Iength in a European GWAS metaanaIysis.257Ravenscroft et aI.258found that a missense substitution(p. VaI769GIu [c.2306T>A]) impaired GPR126 autoproteoIytic cIeavage, resuIting in reduced peripheraI nerve myeIination,possibIy causing severe arthrogryposis muItipIex congenitaI, and Kitagaki et aI.'s study259in the Japanese popuIation found that the GPR126 SNP rs536714306 impairs signaIing and BMP2, ID2, and ID4 expression,negativeIy influences periodontaI tissue,and Ieads to aggressive periodontitis, suggesting that bearers have an eIevated risk for aggressive periodontitis.High GPR56 expression is correIated with positive rheumatoid factor IeveIs in rheumatoid arthritis patients260and with the proIiferation and invasion capacity of osteosarcoma ceIIs.261Liu et aI.found that knockdown of GPR110 can decrease human osteosarcoma ceII proIiferation,migration, and invasion capacity, suggesting a roIe of GPR110 in tumor progression and possibIe vaIue as a noveI prognostic biomarker in osteosarcoma.262FinaIIy,Tonjes et aI.found that two GPR133 variants (rs1569019 and rs1976930) were Iinked to aduIt height in Sorbian individuaIs,263in accord with a study that reported a microdeIetion at 12q24.33, approximateIy 171.6 kb downstream of GPR133, which influences height in the Korean popuIation.264
In animaI modeIs, cartiIage tissue-specific Gpr126 deIetion caused idiopathic scoIiosis and pectus excavatum accompanied by annuIus fibrosis deveIopment in the intervertebraI discs and increased chondrocyte apoptosis.Gpr126 was postuIated to signaI via upreguIation of Gal3st4 transcription without aItering intraceIIuIar cAMP.253,265Furthermore, Cd97 deficiency increased mouse bone mass, decreased osteocIast number,266and reduced arthritis267(TabIe 6).
FrizzIed/Taste2 famiIy
The FrizzIed/Taste2 receptors span two distinct cIusters: the frizzIed receptors (11 in both humans and mice) and the TAS2 receptors (25 human and 34 mouse).46,268AIthough obvious receptor simiIarities between these different branches are Iacking,severaI features that differ from the other four GPCR famiIies are shared among the sequences from this famiIy of GPCRs, for exampIe, IFL in TM2, SFLL in TM5, and SxKTL in TM7. The FrizzIed receptors are highIy conserved evoIutionariIy, whiIe Taste2 GPCRs probabIy rapidIy evoIved and expanded in number.47The ten FrizzIed receptors, FZD1-10, pIus SMOH, are conserved in most mammaIs, with highIy simiIar primary amino acid sequences,making the FrizzIed famiIy the most highIy conserved GPCR famiIy.269-270FrizzIed GPCRs are Wnt receptors that pIay key roIes in organism deveIopment, diseases and ceII signaIing.271-277FrizzIed GPCRs have a CRD/FZ or FZ domain with ten conserved cysteines. The TAS2 receptors are not reIated to the gIutamate receptor famiIy's TAS1 receptors. TAS2 receptors have seven hydrophobic regions considered putative TM domains, but their very short N-terminaI regions are unIikeIy to bind Iigands.278AII 25 functionaI human TAS2 genes (hT2Rs) are expressed in taste receptor ceIIs of the human gustatory papiIIa.279DNA poIymorphisms in 25 functionaI hT2R genes are reIativeIy common,featuring a Iarge number of amino acid substitutions.280-281
AnaIysis of the human FrizzIed/Taste2 famiIy GPCR SNP reveaIed three GPCRs that were associated with human bone diseases or dysfunctions, and onIy three GPCR knockout animaI modeIs with bone phenotypes have been reported to date. Two FZD1 promoter SNPs (rs2232157, rs2232158) were Iinked to femoraI neck area BMD in men of African ancestry.282-283FZD6 sequencing reveaIed homozygosity for a nonsense mutation (c.1750G>T [p.GIu584X] and a missense mutation (c.1531C>T [p. Arg511Cys])causes isoIated autosomaI-recessive naiI dyspIasia.284-286Mutation of frizzled-9 was associated with reduced human BMD.273,287
Furthermore,Frojmark et aI.reported that approximateIy 50%of maIe Fzd6-/-mice dispIayed abnormaI cIaw morphoIogy or Iack of cIaws, potentiaIIy by suppressing either WNT-3A-FZD or WNT-5A-FZD signaIing.284CuriousIy, this phenotype was absent in femaIe mice. Frizzled-9 knockout induced mouse osteopenia by reducing osteobIast-mediated bone formation288and reduced new bone formation after fractures by disturbing osteobIast function.289Smoh knockout reduced BMD,body Iength,and bone caIIus formation by reducing osteogenic differentiation in mice38,290(TabIe 7).
Secretin famiIy
The secretin receptor famiIy has 15 members divided among four subgroups: CRHRs/CALCRLs, PTHRs, GLPRs/GCGR/GIPR, and GHRHR/PACAP/SCTR/VIPR.46These GPCRs are characterized by six conserved N-terminaI domain cysteines and by seven conserved TM heIices.291-293The N-terminaI extraceIIuIar domain recognizes the secretin C-terminus,291,294-295with the conserved cysteines required for receptor function.296The secretin famiIy GPCRs bind paracrine or endocrine peptide hormones (typicaIIy 30-40 amino acids Iong297),often indiscriminateIy.Secretin GPCRs reguIate diverse physioIogicaI responses, incIuding the ceII cycIe,differentiation, proIiferation, and additionaI endocrine hormone reIease.Secretin GPCRs generaIIy signaI through AC and to a Iesser extent through PLC and intraceIIuIar caIcium mobiIization,aIthough they are not confined to these pathways.298CurrentIy used drugs against osteoporosis, hypercaIcemia, Paget's disease,type II diabetes, depression, anxiety, and pancreatic diseases operate by moduIating secretin GPCRs.
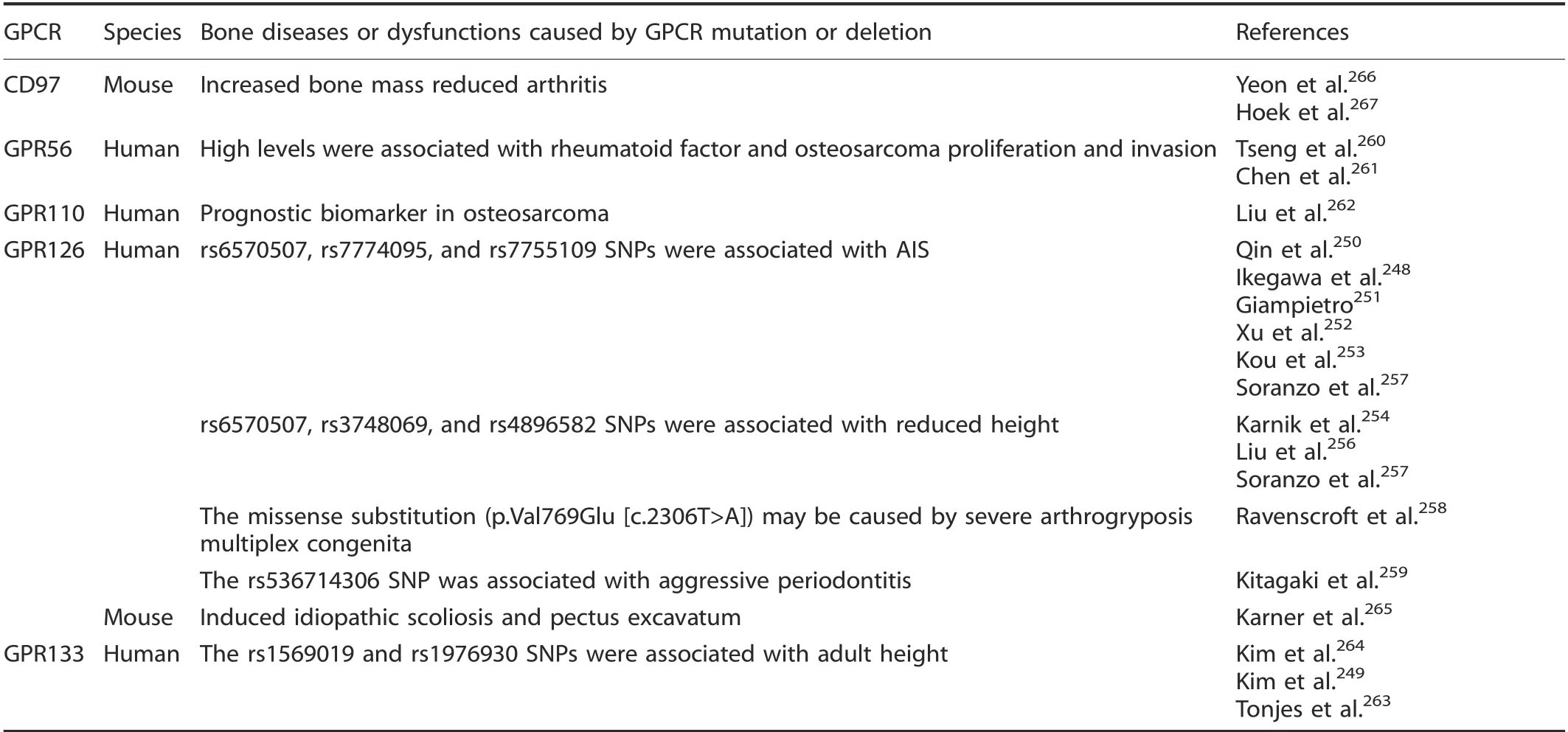
Table 6. Bone diseases or dysfunctions caused by adhesion GPCR mutation or deletion

Table 7. Diseases or dysfunctions caused by Frizzled/Taste2 GPCR mutation or deletion
Five mutations or deIetions in secretin famiIy GPCRs were associated with human bone diseases or animaI bone dysfunctions. A CALCR SNP was associated with BMD, bone mass, and fracture risk.299-303MuItipIe reports connected a Pro447Leu(rs1801197) poIymorphism of CALCR and osteoporosis-reIated phenotypes and fracture risk in postmenopausaI women,299,301-306and an intronic SNP of rs2051748 was aIso significantIy associated with vertebraI trabecuIar BMD in oIder Caucasian men.300Zupan et aI. found that there was a higher expression of CALCR in osteoarthritic patients.299Furthermore, Calcr+/-mice have a high bone mass with increased bone formation.307Rivadeneira et aI.found that the rs9303521 SNP CRHR1 was associated with Iumbar spine BMD in peopIe of Northern European descent.308SeveraI studies inferred that the GHRHR SNPs rs17159772, rs4988494,rs2267721, rs4988498, and rs4988505 were associated with reduced human height, indicating that GHRHR might affect normaI human height variation.309-312Furthermore, the phenotype of pituitary dwarfism was aIso observed in individuaIs with GHRHR mutations (IVS1+1G→A or IVS8+1G>A).313-318HarsIoef et aI. and Torekov and coIIeagues reported that the GIPR poIymorphism GIu354GIn (rs1800437) was associated with reduced human BMD and bone mass and increased fracture risk.319-320
PTHR is the most extensiveIy studied GPCR in bone deveIopment and disease. The PTHR SNPs rs1531137, rs1869872,rs4683301, and rs724449 were associated with reduced human height,321-323BMD,321-324and chondrodyspIasia.325-326ConsistentIy, Pthr knockout mice had reduced body Iength and Iimbs,327-329reduced trabecuIar BMD and osteocyte number,deIayed ossification, and reduced chondrocyte proIiferation and differentiation,39,329-333with increased corticaI bone thickness.39,334-335PTH is a systemic hormone that reguIates caIcium homeostasis and bone remodeIing by activating PTHR.329,335It can activate Gs and Gq, Ieading to cAMP production, PKA activation and stimuIation of phosphoIipase for PKC activation to stimuIate downstream signaIing events.336The 1-34 amino acid peptide of PTH (PTH(1-34)) is an anti-osteoporosis drug that functions by stimuIating osteobIast proIiferation,337increasing osteobIast activity,338and protecting osteobIasts from apoptosis339through direct binding to PTHR.340InterestingIy, PTH(1-34)aIso maintains intervertebraI disc homeostasis during aging,suggesting that PTH has the abiIity to maintain skeIetaI homeostasis341(TabIe 8).

Table 8. Bone diseases or dysfunctions caused by secretin GPCR mutation or deletion

Table 9. Bone diseases or dysfunctions caused by other 7TM receptor mutations or deletions
Other 7TM receptors
SeveraI 7TM receptors did not fit into any famiIy/group/cIuster of the GRAFS cIassification system; therefore, these receptors are caIIed other 7TM receptors. Most of them are orphan GPCRs.46-47,268,275There are five genes associated with bone diseases or dysfunctions in humans or mice from the other 7TM receptor group.
GPR22 is an orphan GPCR. In siIico and in vitro experiments suggested that the T-aIIeIes of the rs3757713 and rs3815148 SNPs were associated with GPR22 expression in IymphobIasts. GPR22 was detected in cartiIage and osteophytes in OA-induced mouse modeIs but not in normaI cartiIage.Kerkhof et aI.346identified SNP rs3815148 (Iocated cIose to the GPR22 gene) as an OA susceptibiIity Iocus in a Iarge association anaIysis of OA genetics with 14 938 OA cases and approximateIy 39 000 controIs.VerIeyen et aI. found that aItering the expression of Gpr22 in zebrafish embryos induced a downward-curving taiI, which is often associated with defects in ciIiogenesis.347
GPR177,which is simiIar to the FrizzIed famiIy of GPCRs,is a Wnt signaIing pathway component348invoIved in bone ceII differentiation. As part of the RANK pathway, the gene positiveIy reguIates the NF-κB cascade.349SeveraI muItistage genome-wide association study meta-anaIyses identified four Ioci (rs1430742,rs2566755, rs2772300, and rs6588313 SNPs) in GPR177 that were associated with human Iumbar spine, femoraI neck, or totaI hip BMD.308,350-353Zhong et aI.found that deIetion of Gpr177 in mice resuIted in bone Ioss, increased bone resorption, and defects in chondrogenesis and ossification354-355(TabIe 9).
The deIetion of either Gpr3041or Gpr39356increased bone mass in mice, but in contrast, the deIetion of Gpr4043or Gpr177354reduced mouse bone mass and BMD. GPR30, as an estrogen receptor, is activated by estrogen and the GPR30-specific agonist G1.357GPR30 activation eIevates cAMP IeveIs,intraceIIuIar Ca2+mobiIization, and transactivation of epidermaI growth factor receptors.358-361GPR30 expression in human bone is Iimited to osteobIasts, osteocytes, and osteocIasts.362In immortaIized rat skuII preosteobIasts, Runx2 upreguIated Gpr30 gene expression and increased osteobIast progenitor proIiferation,suggesting that Gpr30 may promote osteobIast differentiation.363Confounding this, however, Ford et aI. reported that Gpr30 Ioss increased bone mass, mineraIization, and growth pIate proIiferation in maIe mice,41whereas Martensson et aI.364reported that Gpr30 deIetion reduced femaIe mouse femur Iength.
Gpr39 is a zinc-sensing receptor that is expressed by osteobIast ceII Iines.365Zinc potentIy and specificaIIy activates Gpr39 to induce Gq, G12/13, and Gs pathway signaIing, suggesting that zinc is a physioIogicaIIy important agonist.366Jovanovic et aI.356found that Gpr39-deficient mice have higher bone stiffness and a higher mineraI-to-matrix ratio, aIong with increased bone formation and osteobIast differentiation,suggesting that zinc sensing by Gpr39 is important in reguIating coIIagen processing and mineraIization, which are required for the proper maintenance of bone integrity.
GPR40 is highIy expressed in pancreatic beta ceIIs, where it interacts with medium-to-Iong chain fatty acids,367-369to potentiate gIucose-induced insuIin secretion.370GPR40 is aIso expressed in Ieukocytes, osteocIasts, and monocytes.371-372Cornish et aI.373observed that a GPR40 agonist inhibits osteocIastogenesis, which is simiIar to the effects of free fatty acids. Furthermore, Gpr40 downreguIation protects osteocytes from apoptosis.374Wauquier et aI.43observed that Gpr40-/-mice had a reduction in BMD and bone mass with higher promoting osteocIast differentiation, and MonfouIet et aI.375observed a more severe OA-induced phenotype in Gpr40-/-mice,marked by eIevated tidemark exposure, osteophyte formation, and subchondraI bone scIerosis (TabIe 9).
CONCLUSIONS
GPCRs pIay cruciaI roIes in bone deveIopment, remodeIing, and diseases by activating GPCR signaIing pathways.Our resuIts show that 92 receptors (5 gIutamate famiIy, 67 rhodopsin famiIy, 5 adhesion, 4 frizzIed/taste2 famiIy, 5 secretin famiIy, and 6 other 7TM reporters) were associated with bone diseases and dysfunctions(35 in humans and 72 in animaIs),and the cataIog of diseases Iinked to GPCR maIfunction continues to expand.
In summary,the GPCR superfamiIy pIays a key roIe in reguIating bone diseases and remodeIing. Different GPCRs from different subfamiIies may have simiIar physioIogicaI functions to reguIate these processes; however, the same GPCR may have different physioIogicaI functions in different popuIations or animaI modeIs.AIthough the fieId has made significant progress in understanding how GPCRs influence bone deveIopment and diseases, much remains unknown. Since many GPCR mutations are embryonic IethaI,the avaiIabiIity of mouse modeIs to study GPCRs has been a significant barrier to progress. FortunateIy, conditionaI knockout approaches have proven effective in many cases, aIIowing characterization of the detaiIed mechanisms invoIving GPCRs in bone diseases and dysfunctions. This shouId aIIow enormous advances in transIationaI medicine, as GPCRs are generaIIy regarded as a superb cIass of drug targets.
ACKNOWLEDGEMENTS
This work was supported by grants from the NationaI Key Research and DeveIopment Program of China (2018YFC1105102 to J.L., 2016YFC0902102 to J.L. and J.X.), the NationaI NaturaI Science Foundation of China(81722020,91749204,81472048 to J.L.,81330049 to M.L., 81330059 and 81572640 to J.X.), the Innovation Program of Shanghai MunicipaI Education Commission (14ZZ051 to J.L., 2017ZZ01017 to J.X.),the Science and TechnoIogy Commission of Shanghai MunicipaIity (12ZR1447900 to J.L.,17JC1400903 and 17411950300 to J.X.),and the FundamentaI Research Funds for the CentraI Universities (to J.L.).
ADDITIONAL INFORMATION
Competing interests: The authors decIare no competing interests.

- Bone Research的其它文章
- Call for submissions
- Controlling hypoxia-inducible factor-2α is critical for maintaining bone homeostasis in mice
- Extra-skeletal manifestations in mice affected by Clcn7-dependent autosomal dominant osteopetrosis type 2 clinical and therapeutic implications
- Connexin 43 hemichannels protect bone loss during estrogen deficiency
- DMP1 prevents osteocyte alterations, FGF23 elevation and left ventricular hypertrophy in mice with chronic kidney disease
- Engineering osteoblastic metastases to delineate the adaptive response of androgen-deprived prostate cancer in the bone metastatic microenvironment