
Roles of cell fusion, hybridization and polyploid cell formation in cancer metastasis
2020-06-19 06:35IvanShaboJoarSvanvikAnnelieLindstrTanguyLechertierSaraTrabuloJamesHulitTimSpareyJohnPawelek
Ivan Shabo, Joar Svanvik, Annelie Lindstr?m, Tanguy Lechertier, Sara Trabulo, James Hulit, Tim Sparey,John Pawelek
Abstract Cell-cell fusion is a normal biological process playing essential roles in organ formation and tissue differentiation, repair and regeneration. Through cell fusion somatic cells undergo rapid nuclear reprogramming and epigenetic modifications to form hybrid cells with new genetic and phenotypic properties at a rate exceeding that achievable by random mutations. Factors that stimulate cell fusion are inflammation and hypoxia. Fusion of cancer cells with non-neoplastic cells facilitates several malignancy-related cell phenotypes, e.g., reprogramming of somatic cell into induced pluripotent stem cells and epithelial to mesenchymal transition. There is now considerable in vitro, in vivo and clinical evidence that fusion of cancer cells with motile leucocytes such as macrophages plays a major role in cancer metastasis. Of the many changes in cancer cells after hybridizing with leucocytes, it is notable that hybrids acquire resistance to chemo- and radiation therapy. One phenomenon that has been largely overlooked yet plays a role in these processes is polyploidization. Regardless of the mechanism of polyploid cell formation, it happens in response to genotoxic stresses and enhances a cancer cell’s ability to survive. Here we summarize the recent progress in research of cell fusion and with a focus on an important role for polyploid cells in cancer metastasis. In addition, we discuss the clinical evidence and the importance of cell fusion and polyploidization in solid tumors.
Key words: Cell fusion; Hybrid formation; Polyploidization; Macrophage; Cancer progression; Oncologic treatment resistance
INTRODUCTION
Cell fusion is a common biological process that produces viable cells and plays a major role in mammalian tissue development and differentiation. Cell fusion is essential during embryogenesis and morphogenesis,e.g., when trophoblasts in placenta fuse to form syncytiotrophoblasts, in developing of skeletal muscle that arises from the fusion of mesodermal cells[1], and when osteoclasts and multinucleated giant cells are generated from the fusion of macrophages[2,3]. Cell fusion also plays a role in tissue repair and regeneration,e.g., in liver, heart and intestine[4-6]. Through cell fusion, somatic cells undergo nuclear reprogramming and epigenetic modifications to form pluripotent hybrid cells. Cell fusion can result in rapid modifications of the genetic and epigenetic programs of cells, generating cells with new properties at a rate exceeding that achievable by random mutations[6-8].
Cell fusion may result in two forms of hybrids: Heterokaryons or synkaryons.When bi- or multinucleated hybrids are generated (heterokaryons), the parental genomes are located in different nuclei, segregated from one another. These hybrids are capable of cell divisions resulting in daughter cells expressing both parental sets of chromosomes in a single nucleus[9,10]. Hybrids with parental genomes mixed in a single nucleus are called synkaryons[11-14]. In culture, fusion events depend on the cell density, the cell ratio of the parental populations and their microenvironment[15].
CELL FUSION, POLYPLOIDIZATION AND CANCER
Polyploid cells contain more than two basic sets of chromosomes. Polyploidy is a natural phenomenon that contributes to tissue differentiation, normal organogenesis and tissue repair[16,17]. It appears that most polyploid cells in mammals are formed through cell fusion[18], but also through abnormal cell division such as endoreplication,endomitosis and failed cytokinesis after completion of mitosis[19-21]. The formation of multinucleated fusion hybrids may allow genetic complementation capable of rescuing loss of gene function after chemotherapeutic or radiation induced DNA damage[22-24]. Formation of polyploid cells with a selective advantage may serve as a cell survival mechanism[25].
In 1911, a German professor, Otto Aichel, proposed that fusion of cancer cells with leucocytes produces hybrids with a metastatic phenotype. A century later, this prescient insight has proven to be correct in many studies. Indeed, leucocyte-cancer cell fusion causes cellular reprogramming and generates new clones of hybrids at least some of which acquire the ability for chemotactic migration by combining the epigenetic program of the leucocyte with the uncontrolled cell division of the cancer cell[8,26,27](Figure 1). Tumour cells are fusogenic and fuse with other cancer cells and non-neoplastic cells in the tumour stroma. Spontaneous fusion between cancer cells is a well-documented phenomenon in solid tumors and generates heterogeneous subpopulations of tumor cells[28-31]. Several studies have shown that fusion of cancer cells with non-neoplastic cells facilitates several malignancy-related cell phenotypes,e.g., reprogramming of somatic cell into induced pluripotent stem cells and epithelial to mesenchymal transition (EMT) (Figure 1). These processes also produce cellular polyploidy.
Polyploid giant cancer cells (PGCCs) are cells with multiple nuclei or a single giant nucleus containing multiple sets of chromosomes. Compared to regular diploid cells,PGCCs can be 10 to 20 times larger in size and have tetraploid or greater (≥ 4C) DNA content[32]. The mechanism leading to formation of PGCC may have similarities to wound repair and tissue regeneration, each of which utilizes or depends on cell fusion and endomitotic and endoreplication mechanisms. Regardless of the mechanism of polyploid cells formation, it happens in response to genotoxic stresses such as those occurring in hypoxic/necrotic regions of the tumor and during chemo- and radiotherapy. Polyploidy then enhances a cancer cell’s ability to survive. Even though the formation of hybrids between cancer cells and bone marrow derived cells(BMDCs) most likely occurs stochastically[26], disease progression and standard of care treatments such as chemo- and radiotherapy, can act as driving forces that may increase the frequency of hybridization events[33,34]. The basis of this increase may be due to increased inflammation and/or an aberrant wound and damage response mechanism leading to the formation of PGCC[35,36].
Aneuploidy is a hallmark of cancer and is proposed to have a fundamental role during tumour initiation and progression. Approximately 90% of solid tumors and 75% of hematopoietic malignancies have abnormal chromosome numbers[37,38].Centrosome aberrations are suggested as one mechanism that causes the formation of aneuploid genomes. An important evolutionary feature of polyploid cancer cells is the generation of aneuploid clones during the reversal of the polyploid state, which is a chaotic process with many genomic translocations, amplifications and deletions occurring during creation of progeny[39]. The presence of centrosome aberrations in polyploid cancer cells suggests that cell fusion and the formation of polyploid cancer cells may be strong contributors to aneuploidy[40,41].
Cell fusion and cancer-stem cells
Cancer stem cells (CSCs) are subpopulations of tumour cells with stem cell-like traits.These traits include high plasticity and the capacity for self-renewal through asymmetric division. CSCs sustain tumourigenesis and generate diverse progeny with the ability to remain dormant while demonstrating resistance to conventional cancer therapeutics[42]. The stem cell theory in cancer, however, is debated due to controversy about evidence supporting the origins of CSCs, their differentiation and dedifferentiation, genetic heterogeneity, symmetric and asymmetric concepts of cellular division, and clonal evolution[43,44].
Cell fusion could be a mechanism to generate CSCs (Figure 2). Gaucket al[45]showed that spontaneous fusion of human breast epithelial cells and human breast cancer cells can give rise to hybrid cells that possess CSC properties with significantly increased colony forming capacity compared to the maternal epithelial cells. In a polyethylene glycol mediated fusion experiment, Flaszaet al[46]demonstrated that murine P19 and human NTERA2/D1 embryonal carcinoma hybrid cells displayed heterogeneity in cellular morphology and gene expression. The hybrids expressed stemness factors octamer-binding transcription factor 4, homebox protein Nanog and Sex Determining Region Y-Box 2, indicating the activation of endogenous human markers of pluripotency. In another example, spontaneously formed heterotypic hybrids between mesenchymal stem cells and lung cancer cells expressed the stem cell marker prominin-1, which was increased 30-fold in hybrids cells compared to their maternal lung cancer cells. The hybrids also exhibited increased expression of the transcription factors octamer-binding transcription factor 4, Aldehyde dehydrogenase 1, B-lymphoma Mo-MLV insertion region 1, and Sex Determining Region Y-Box 2, suggestive of a stem cell-like phenotype[47].
Cell fusion causes chromosomal instability, tumour heterogeneity and DNA exchange
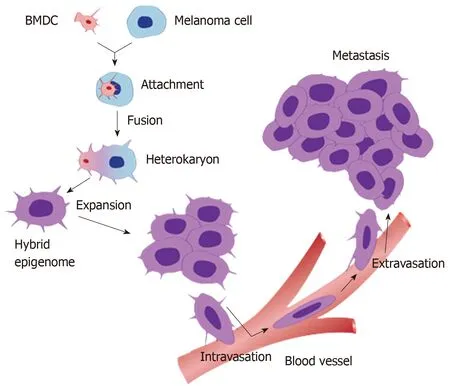
Figure 1 A schematic diagram of the process of cell fusion, hybrid formation and metastasis. A motile bone marrow derived cells (red) such as a macrophage or stem cell is drawn to a cancer cell (blue). The outer cell membranes of the two cells become attached. Fusion occurs with the formation of a bi-nucleated heterokaryon having a nucleus from each of the fusion partners. The heterokaryon goes through genomic hybridization creating a cancer cell-bone marrow derived cells hybrid with co-expressed epigenomes, conferring deregulated cell division and metastatic competence to the hybrid. BMDC: Bone marrow derived cell.
Most malignant tumors are polyclonal. Clonal heterogeneity may be caused by oncogenic mutations in single cells, epigenetic modulations[48], and cell fusion[49].Chromothripsis, massive genomic rearrangements[50], is defined as a single catastrophic event in the development of cancer[51]. In oral squamous cell carcinoma cell lines in culture, irradiation predisposes to cells chromothripsis. The finding of fragmented DNA in aneuploid hybrid cells is an indicator of chromothripsis[52].
Cell fusion may be homotypic between the same types of cells in the tissue or heterotypicbetween different cell types like epithelial cells and macrophages.Heterotypic fusion can cause multiple changes in gene expression profiles in the resultant hybrids[10]. Clonal heterogeneity patterns within primary tumors are often similar to those of distant metastases with similar gene expression profiles. Using a Cre-loxP model system, Searleset al[53]showed that Cre transfer occurred between cancer and non-cancer cells both in cell cultures and in mice. The rapid transfer of Cre could not be explained by extracellular vesicles but rather by cell fusion.
Cell fusion, cancer and EMT
In order to form metastases, tumour cells need to navigate through a series of obstacles that require a variety of cellular functions and abilities that were absent in the transformed cells of origin. The functions include an invasive escape from the tumour and intravasation into blood or lymphatic vessels. All steps of the metastatic cascade require an ability to overcome the induction of cell death. To escape the circulation, tumour cells need to adhere to the vessel wall and undergo extravasation into other tissues. Once in the tissue, cell growth is required to form metastasis. One mechanism put forth to explain the changes required to perform these functions is EMT. This model explains how neoplastic cells may gain a migratory and invasive phenotype allowing them to escape from the primary tumour. Many studies have identified a subset of embryonic-like transcription factors, such as zinc finger protein SNAI1 and basic helix-loop-helix factor Twist, that form the basis of a gene expression program that drives the transitional change of the phenotype. An alternative mechanism is that cancer-mesenchymal cell fusions generate hybrids that gain the genetic, phenotypic and functional properties of both maternal cells. Xuet al[54]showed in anin vivonon-obese diabetic/severe combined immunodeficiency mouse model that fusion of mesenchymal stem cells with non-small cell lung cancer cells results in hybrids that express both epithelial and mesenchymal markers with increased migratory and invasive capabilities compared to their maternal cancer cells.
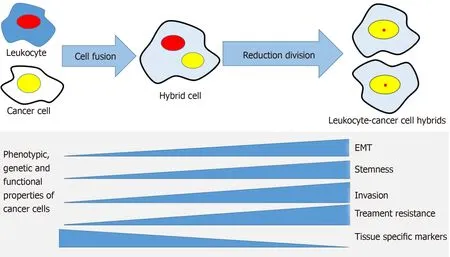
Figure 2 The cell fusion theory in relation to cancer progression mechanisms. A cancer cell and a leukocyte form a hybrid that acquires genetic, phenotypic and functional properties from both maternal cells. The hybrid cells develop properties associated with cancer metastasis, such as epithelial mesenchymal transition,stemness, invasiveness, treatment resistance and may lose some of the maternal cancer cell's tissue-specific phenotype. EMT: Epithelial to mesenchymal transition.
In studies by Zhanget al[55], analysis of polyploidy giant cells (referred to by the authors as “PGCC”)in colorectal cancer revealed a strong association with the presence of lymph node metastasis. Potentially the PGCC were responsible for metastasis as a subset of “budding” daughter cells showed a greater migratory and invasive phenotype and expressed the EMT-related proteins Twist and Snail.Similarly, PGCCs induced by the hypoxia mimetic cobalt chloride were capable of generating small diploid cell progeny that also displayed higher levels of EMT related protein expression including vimentin and N-cadherin. These daughter cells had a more invasive phenotype compared to the parental cell type. Importantly, the authors showed that patient samples from breast tumours and metastasis had an increased number of PGCCs with vimentin and N-cadherin expression compared with patient breast tumour samples with no metastasis[56], indicating a higher metastatic potential of the progeny from the PGCCs.
PGCCs and mitochondrial function
PGCCs form under a diverse set of stimuli as they are found within and adjacent to necrotic regions of tumours, driven by conditions of hypoxia, nutrient deprivation(starvation) and low pH. Individually these stimuli have been shown to induce PGCCin vitro. Importantly, oxidative stress is the common feature that links these stimuli.Chemotherapies and radiotherapy which can dramatically induce the transformation to PGCCs also generate oxidative stress generated through excess reactive oxygen species (ROS). Mitochondria are the main generator of ROS and may also act as“sensors” to trigger the PGCC transformation which form and exist under conditions of high oxidative stress[57-60]. Rohet al[57]showed that PGCCs have increased mitochondrial content as well as elevated levels of ROS. The authors found a correlation between the high ROS levels characteristic of the polyploid cells together with taxol resistance that could be reversed with the use of antioxidants. Similarly, the ROS-producing agent plumbagin induced features of PGCCs on prostate cancer cells,in line with the idea that polyploidy provides a survival advantage to cells that are exposed to high levels of ROS. The authors suggested that besides impacting cancer cell resistance to therapy, the polyploid state could also contribute to the generation of CSCs in response to stress[59]. In the tumour microenvironment, where growth conditions deteriorate and oxidative stress increases with progression[61], PGCC are induced at higher rates[33,62]. Even the number of circulating hybrid cancer cells is increased as shown in liquid biopsies[63-66].
How cell fusion promotes tumour progression
Cell fusion contributes to tumour progression not only through alteration of the composition and biology of tumour cellsper se, but also by modifying the tumour microenvironment. Hybridization of cancer cells withe.g., BMDCs, generates hybrids with a significantly faster growth ratein vivo[67,68]and enhanced abilities of colony formation[68], cell migration and invasion[47]. Thus, cell fusion alters the biological behaviour of a tumour through the development of new metastatic tumour cell sets with growth-promoting properties, contributing to enhanced tumour growth and metastasis[69]. Similar observations have been reported in several other studies over the past three decades[14,70-73].
Cell fusion also contributes to neo-angiogenesis and thus facilitates hematogenic and lymphatogenic intravasation of tumour cells. Recently, Shenet al[74]demonstrated that dendritic cells can fuse with endothelial progenitor cells and generate hybrids with a significantly faster growth rate, while also producing a greater number of micro-vessels compared to their maternal endothelial progenitor cell. Busundet al[75]showed thatin vivogrowth of cultured tumours consisting of Metha-A sarcoma cell/macrophage hybrids had a significantly higher intratumoural microvessel density and maturation compared to tumours from maternal Metha A sarcoma cells alone. The hybrids released significantly higher amounts of angiogenic peptides, such vascular endothelial growth factor (VEGF), compared to both maternal macrophages and cancer cells. In renal tissues of individuals with gender-mismatched transplants who had transplant rejection and chronic inflammation, Kerjaschkiet al[76]provided clinical evidence that BMDCs, presumably macrophages, function as progenitors of lymphatic endothelial cells, and contribute to lymphangiogenesis by incorporating into the new lymphatic vessel. Moreover, the macrophage might differentiate to VEGF producing cells that drive the division of endothelial cells[77,78].
MACROPHAGES AS FUSION PARTNERS IN CANCER
Macrophages are a heterogeneous population of cells derived from monocytes.During embryogenesis, they appear first in the yolk sac, then in the liver, and finally in bone marrow. Large populations of tissue macrophages exist in the small intestine,liver (Kupffer cells), and lungs. Blood monocytes arise in the marrow from precursor cells (monoblasts) and enter inflamed or infected tissues, where they may mature into macrophages and increase the resident macrophage populations. Monocytes may also mature into dendritic cells presenting antigens to T cells. Fusion is an important function of macrophages and results in the formation of osteoclasts and multinucleated giant cells[3].
Macrophages show two different polarization states, M1 and M2, in response to different signals in the microenvironment. M1 macrophages are pro-inflammatory and characterized by the release of inflammatory cytokines and microbicidal/tumoricidal activity. M2-macrophages have an immunosuppressive phenotype and are polarized by anti-inflammatory molecules such as Interleukin (IL)-4, IL-13, and IL-10, apoptotic cells, and immune complexes. M2 macrophages release antiinflammatory cytokines and have scavenging potentials as well as supporting angiogenesis and tissue repair. Monocyte/macrophage cells are important for tumour cell migration, invasion and metastasis. Tumour associated macrophages (TAM)represent the M2-type and promote tumor progression[79-83].
Monocytes are actively recruited to the tumour stroma, and high infiltration of TAMs in many tumour types correlate with lymph node involvement and distant metastases[84]. Inhibition of macrophage infiltration in tumours may suppress metastasis[85,86]. The clinical significance of macrophage infiltration in tumour stroma,however, is still controversial. High infiltration of TAMs is correlated to poor prognosis in breast, prostatic, ovarian and cervical carcinoma[87]. In colorectal cancer,there are conflicting data of the clinical significance of macrophage infiltration, but several studies show that low macrophage density in tumour stroma is associated with an unfavourable prognosis[87-89].
TAMs contribute to angiogenesis, lymphangiogenesis and tumour progression by expressing pro-angiogenic growth factors such as matrix metalloproteinase-12, IL-1,VEGF, and IL-8. Clinical studies have shown that increased infiltration of TAMs in solid tumours is associated with high micro-vessel density and poor prognosis. These data are particularly strong for hormone-dependent cancers, such as breast cancer[87,90].
INFLAMMATION AND CELL FUSION
The mechanisms causing cell fusion and polyploidization in tumours have close similarities to wound repair and tissue regeneration in non-transformed tissues.Under normal conditions, fusion events are exceptionally rare but increase dramatically in pathological conditions such as after tissue injury and during inflammation. Nygrenet al[34]showed that BMDC contribute to the formation of stable and reprogrammed fusion-hybrids in liver, heart and myocardium during tissue repair. Interestingly, the authors observed that despite an attraction of fusogenic blood cells to these tissues following injury-induced inflammation, the cell fusion was restricted to a subset of cells implicated in syncytia formation during development.Johanssonet al[91]reported in a parabiotic experimental mouse-model that chronic inflammation, induced by idiopathic ulcerative dermatitis and autoimmune encephalitis, could increase myelomonocytic cells in peripheral blood and consequently significantly increase (10–100-fold higher) heterokaryon formation of BMDCs with Purkinje neurons.
HYPOXIA/TISSUE STARVATION AND CELL FUSION
The tumour microenvironment is usually characterized by poor vascularization,resulting in hypoxia and deficient access to vital nutrients and elimination of metabolic by-products. Necrosis is common in the deeper parts of the tumour. Despite these seemingly harsh conditions, cells that subsist in this environment have been linked to a more malignant phenotype with stem cell-like properties[92,93].
As mentioned above, cell fusion is induced by hypoxia and apoptosis. Noubissiet al[94]used bimolecular fluorescence complementation to detectin vitrospontaneous fusion events between co-cultured multipotent stem/stromal cells (mSSC) and either human breast epithelial cells (MCF-10a) or breast cancer cell lines (T47D, MDA-MB-231 and MCF-7). The co-cultures were grown in hypoxic condition (2% O2compared to standard tissue culture conditions of 21% O2) and it was found that hypoxia stimulated a significant increase in fusion between the mSSC and the T47D or MCF-7 non-metastatic breast cancer cells. Hypoxia had, however, no significant impact on the fusion ability of MDA-MB-231 metastatic cancer cells. The authors suggest that hypoxia might promote fusion of non-metastatic cancer cells and therefore enables metastasis, an event that perhaps is not advantageous for the already metastatic MDA-MB-231 cells. It was found also that apoptosis was enhanced by hypoxia in T47D and MCF7 non-metastatic cancer cells. The fusion events were increased if the cell co-cultures were supplemented with apoptotic T47D cells in both normoxic and hypoxic conditions, indicating that cell fusion could be stimulated by apoptosis independently of hypoxia. Melzeret al[95]reported similarin vitroobservations and found that cell fusion events varied dramatically between benign or malignant breast cells when used as fusion partners for mSSC. Co-cultures of mSSC with MCF-10a revealed increased fusion events up to 10-fold (> 2%) compared with co-cultures with MDA-MB-231 (0.2%) or with MCF-7 (0.1%). In line with the observations that inflammation can induce cell fusion, the author stimulated the cell co-cultures with pro-inflammatory cytokine TNF-α and found significant hybrid cell formation compared to non-treated cells.
Using different cancer cell lines treated with the hypoxia mimetic cobalt chloride to induce polyploidization, Zhanget al[55]showed that the formation of PGCCs could occur either through cell-fusion or endoreplication. The cells had increased size,developed enlarged nuclei and could survive for extended periods of time, while cells with normal morphology could be selectively eliminated. The PGCCs displayed CSC markers and properties and could therefore be potentially more tumourigenic[96].
CLINICAL SIGNIFICANCE OF CELL FUSION AND POLYPLOIDIZATION IN CANCER
PGCCs are found in most cancers and correlate to poor survival[97-100]. Reports of mono and multinucleated cancer cells can be found in the literature dating back to the early 20thcentury[101-103]. PGCCs are also described as “multi-nucleated giant cells”, “tumour budding”, “micropapillary”, and “osteoclast-like giant cells”. Across many cancer types, when present in high numbers, PGCCs of epithelial-origin show highly malignant characteristics, including chemo-resistance, with short patient survival and are designated with World Health Organization sub-classification status. These rare cancers include, giant cell carcinoma of the lung (a sub-classification of the large cell/sarcomatoid carcinomas of the lung) and pleomorphic carcinoma of the lung[104-106]. Although rare, these cancers provide important insights into the highly malignant nature of the PGCCs. Patient survival is typically worse than in those with the non-PGCC cancer component due to poor or refractory responses to standard treatments, short relapse intervals and enhanced metastatic spread[32,56,97,99].
PGCC formation has been demonstrated to increase during tumour progression[55,56,99,100], as late stage cancers have elevated numbers of PGCCs. It might be predicted that through increased PGCC formation in late stage disease, increased numbers of heterotypic fusion events also occur. Recent studies on liquid biopsies show the presence of hybrid-like expression in multinucleated giant cells. This phenotype is shown to correlate with malignancy and poor survival[66,107-109].
Several studies have shown that after standard of care treatments treatments with radiation[33,35,110], chemotherapy[40]or targeted therapy[111], PGCC can be formed using cell fusion as a mechanism. The PGCCs can overcome the treatment-induced damage and produce progeny that are resistant to treatments to which the cancer cells are normally sensitive. This has been demonstrated for both irradiation and chemotherapy and may be an important factor in disease relapse and patient outcome[40,112-114].
PGCCs have been shown in many tumour types to be associated with increased therapy resistance and poor survival[104,106,115]. For example, in a syngeneic rat tumour model, Puiget al[116]showed that despite an initial shrinkage of tumours in response to cisplatin treatment, tumour cells enter a latent phase from which they are eventually able to escape and resume growth. These cells survived for weeks and while some eventually died, others were able to undergo a reversal of polyploidization giving rise to new colonies of diploid cells, which were more resistant to cytotoxic drugs and were responsible for tumour relapse. Additionally, a single large multinucleated cell isolated from the murine fibrosarcoma cell line UV-2237 could produce tumours in mice. These large multinucleated cells were also more resistant to doxorubicin suggesting that they could be driving the relapse[117].
Clinical evidence of cell fusion
It has been well demonstrated that leucocyte-cancer cell fusion produces hybrid cells that express genetic and phenotypic characteristics of both maternal cells[71,118].Clinically, it is difficult to detect or genetically confirm fusion events because the genetic content of maternal cells and any hybrids have the same origin. The expression of tissue specific proteins by tumour cells and other fusion cell partners,like TAMs, may, however, constitute surrogate markers that could be used for detecting the presence of fusion events in tumour tissue from clinical patient material.
CD163 is a macrophage specific trans-membrane scavenger receptor and its presence indicates that the cells have an M2-macrophage phenotype[119]. Macrophage traits in cancer cells, exemplified by CD163 expression, have been reported for several types of tumours,e.g., renal cell[120], breast[121], colorectal[122,123]and bladder[124,125]cancers.Based on the cell fusion theory, the macrophage phenotype in cancer cells is suggested to be caused by fusion between TAMs and tumour cells[71,126,127].
In anin vitromodel, Shaboet al[126]showed that cancer cells did not acquire a macrophage phenotype by paracrine interaction between macrophages and MCF-7 breast cancer cells. In contrast, macrophage/MCF-7 hybrids (generatedviaspontaneous cell fusion) expressed macrophage-like markers, CD163 and the panleucocyte marker CD45. The hybrids also showed genetic characteristics from both parent cells. Powelet al[71,128]providedin vivoevidence of fusion between circulating BMDCs and cancer cells during tumourigenesis, demonstrating that macrophages were cellular partners in this process. Silket al[118]showed similarin vivocharacteristics in human intestinal epithelium. These studies clearly support the many observations that macrophage traits in cancer cells are explained by fusion between tumour cells and TAMs[4,67,75].
Shaboet al[121]reported that CD163 expression by tumour cells in breast cancer was seen in 48% of a cohort of 133 female patients. The patients with CD163-positive tumours had reduced recurrence-free survival times. Further, CD163 expression by cancer cells was more common in advanced cancers. Epithelial cells in normal breast tissue showed no expression of CD163. In a similar study, the same research group reported CD163 expression by tumour cells in 23% of 139 patients with rectal cancer.Again, CD163 expression was not seen in any of the non-cancerous areas from adjacent or distant rectal tissue. CD163 expression by tumour cells was associated with earlier local recurrence and shorter cancer specific survival, and inversely correlated to apoptosis. Notably, the expression of CD163 by cancer cells was more common (31%) in tumours from patients treated with preoperative radiotherapy compared to those not treated (17%).
The expression of macrophage traits by cancer cells was proportional to intratumoural macrophage density indicating that increased recruitment and infiltration of macrophages in tumour tissue may result in higher rates of fusion between macrophages and cancer cells in tumour stroma[122,124]. The frequency of cell fusion eventsin vivowas as high as 1% in experimental tumour models[27]. The fusion efficiency increased proportionally to the presence of inflammation[91]and the metastatic potential of tumour cells[129]. Macrophage-cancer cell hybrids are generated spontaneously in cultured breast cancer cells at an average rate of 2 % and are able to survive cell culture for several weeks[126]. Del Monte demonstrated that one gram of tumour contained some 108tumour cells[130]. Based on this calculation, a rate of 1%-2 %frequency of cell fusion events means that each gram of solid tumour may contain as many as 1-2 million hybrids. Although the proportion of hybrids may be small in relation to the total tumour mass, the spontaneity of cell fusion and the survival of the hybrids may generate deadly derivative clones with important clinical implications.Garvinet al[131]calculated the proportion of cancer cells expressing CD163 in 83 patients with breast cancer and found that the number of CD163-positive cells in all breast tumors studies averaged 9% (range 0%-41%). CD163-expression of > 15% of cancer cells was associated with breast cancer-related death (P= 0.02). The authors also reported that the mean number of cancer cells expressing CD163 was positively associated with mitotic index supporting a connection between fusion events and the density of TAMs seen in tumours. It is likely that the plasticity of reprogrammed cellular phenotypes originating in hybrids may form both dominating as well as volatile clones in the tumour environment.
Through cell fusion cancer cells acquire new phenotypes but may also lose other traits that are specific for their tissue origin, a process known as de-differentiation.These traits are essential in the clinical assessment,e.g., the estrogen receptor (ER)pathway is involved in cell growth and regulation of breast cancer cells. Moreover,estrogen is a potent breast mitogen and ER-inhibitors and estrogen-producing enzymes (aromatases) are well-established, effective therapies[132]. ER is downregulated in progeny cells generated by fusion between macrophages and breast cancer cells[133]. In immuno-histochemical studies of biopsies, macrophage traits in cancer cells (indicating fusion events) was associated with ER-negative tumours[121,131].These observations have clinical relevance as down-regulation of ER in breast cancer cells will change the pathologic staging and the treatment options for the patients.
Cell fusion pathways as diagnostic and therapeutic targets
Clinical investigations of tumour biology with a focus on cell fusion as an underlying mechanism are limited, probably because this theory has been difficult to investigate and has not been firmly established as other topics, such as the cancer mutation theory. In light of findings on the role of cell fusion in tumour biology reported over the past 30 years, we believe that cell fusion pathways might constitute a target in cancer diagnostics and treatment. As discussed previously, cell fusion contributes to tumour progression by generating new cancer cell clones with enhanced metastatic properties. Morphologically, hybrid cells have similar appearance to their maternal cancer cells. Hence, the malignant potential of tumours might be underestimated if hybrid cells are not detected during clinical histopathological assessment. For example, Busundet al[75]showed thatin vivotumours consisting of Metha-A sarcoma/macrophage hybrids had the similar histopathological morphology as tumours consisting of maternal Metha-A sarcoma cells. The tumours consisting of hybrid cells had however greater growth rate, metastatic ability and vascular density;tumour characteristics that have prognostic significance in a clinical context.
Consistent within vivofindings, the expression of macrophage traits as surrogate marker for macrophage-cancer cell fusion[4,71,118,134]by cancer cells in clinical tumour material is associated with advanced tumour stage and poor prognosis[120-125,135,136],indicating the importance of identifying and validating histopathological markers,such as macrophage-specific marker CD163, to detect fusion events in clinical tumour material. Accumulating evidence suggests that cell fusion results in the development of stem cell properties and resistance to oncological treatment[33,115,127,137-140]. Cell fusion may have a predictive value in cancer treatment and it should be verified by clinical investigations.
Cell fusion might constitute a therapy target in cancer and can be counteracted by several strategies. Tentatively, inhibition of the cell fusion process or infiltration of cancer cell fusion partners, such as macrophages, might be possible treatment targets.Inhibition of macrophage infiltration into tumour stroma might reduce the frequency of macrophage-cancer cell fusion. Macrophage depletion might also reduce other macrophage related tumour promoting mechanism such as neo-angiogenesis.Although these data are not related to macrophage-cancer cell fusion, several studies show that macrophage depletion in tumour stroma is associated with inhibition of tumour progression[141-144]. For example, Griesmannet al[145]showed in an experimental mouse model that treatment with liposomal clondronate decreased macrophage infiltration in several organs and resulted in significant reduction of liver and pulmonary metastasis of pancreatic cancer, independently of the presence of an endogenous primary tumour.
RATIONALE
Using histochemical markers along with genetic analyses it is now clear that cell fusion and hybrid formation are associated with metastasis and poor patient survival.There is an association of polyploidy, produced by leucocyte-cancer cell fusion, with therapy resistance. We may glimpse the engine that drives metastasis (Figures 1 and 2). This information opens many potential targets for the development of new therapies: (1) Inhibition of the fusion process itself regarding events such as membrane attachment and heterokaryon formation; (2) Inhibition of the hybridization processes involving integration of parental fusion partner genes into hybrid genomes;and (3) Prevention of post-hybridization events involving activation of genes that control cell migration, chemotaxis, intravasation, extravasation, and migration to lymph nodes and distant metastases.
CONCLUSION
Cell fusion is a normal biological process that is essential during embryogenesis and morphogenesis. Accumulating evidence indicates that fusion between leukocytes and cancer cells occur in solid tumors and may contribute to tumor progression. These data provide new insights into the role of leukocytes, such as macrophages, in tumor biology and cell fusion as a potential mechanism in tumor metastasis and the development of resistance to oncologic treatment.
 World Journal of Clinical Oncology2020年3期
World Journal of Clinical Oncology2020年3期
- World Journal of Clinical Oncology的其它文章
- Thrombocytopenia with multiple splenic lesions - histiocytic sarcoma of the spleen without splenomegaly: A case report
- What factors influence patient experience in orthopedic oncology office visits?
- Efficacy, patterns of use and cost of Pertuzumab in the treatment of HER2+ metastatic breast cancer in Singapore: The National Cancer Centre Singapore experience
- Assessment methods and services for older people with cancer in the United Kingdom
- Glycoconjugation: An approach to cancer therapeutics