
IC4R-2.0: Rice Genome Reannotation Using Massive RNA-seq Data
2020-11-06 01:04:04JianSangDongZouZhennanWangFanWangYuanshengZhangLinXiaZhaohuaLiLinaMaMengweiLiBingxiangXuXiaonanLiuShuangyangWuLinLiuGuangyiNiuManLiYingfengLuoSongnianHuLiliHaoZhangZhang
Jian Sang, Dong Zou, Zhennan Wang, Fan Wang,Yuansheng Zhang,3, Lin Xia,3, Zhaohua Li,3, Lina Ma, Mengwei Li,3,Bingxiang Xu, Xiaonan Liu,3, Shuangyang Wu, Lin Liu,3,Guangyi Niu,3, Man Li,3, Yingfeng Luo, Songnian Hu,??,*, Lili Hao,*,#,Zhang Zhang,3,*
1 CAS Key Laboratory of Genome Sciences and Information, Beijing Institute of Genomics, Chinese Academy of Sciences,Beijing 100101, China
2 National Genomics Data Center, Beijing Institute of Genomics, Chinese Academy of Sciences, Beijing 100101, China
3 University of Chinese Academy of Sciences, Beijing 100049, China
4 State Key Laboratory of Integrated Management of Pest Insects and Rodents, Institute of Zoology, Chinese Academy of Sciences, Beijing 100101, China
KEYWORDS Genome reannotation;IC4R;Rice;RNA-seq;Gene model
Abstract Genome reannotationaims for complete and accurate characterization ofgene models and thus is of critical significance for in-depth exploration of gene function.Although the availability of massiveRNA-seqdata provides great opportunities for gene model refinement, few efforts have been made to adopt these precious data inrice genome reannotation. Here we reannotate the rice(Oryza sativa L.ssp.japonica)genome based on integration of large-scale RNA-seq data and release a new annotation system IC4R-2.0.In general,IC4R-2.0 significantly improves the completeness of gene structure, identifies a number of novel genes, and integrates a variety of functional annotations.Furthermore,long non-coding RNAs(lncRNAs)and circular RNAs(circRNAs)are systematically characterized in the rice genome.Performance evaluation shows that compared to previous annotation systems, IC4R-2.0 achieves higher integrity and quality, primarily attributable to massive RNA-seq data applied in genome annotation. Consequently, we incorporate the improved annotations into the Information Commons for Rice(IC4R),a database integrating multiple omics data of rice, and accordingly update IC4R by providing more user-friendly web interfaces and implementing a series of practical online tools.Together,the updated IC4R,which is equipped with the improved annotations, bears great promise for comparative and functional genomic studies in rice and other monocotyledonous species. The IC4R-2.0 annotation system and related resources are freely accessible at http://ic4r.org/.
Introduction
As a major crop for more than 7000 years, rice is one of the most important staple food feeding a large number of people throughout the world, with vital significance for global food security.Possessing a relatively small genome and high genetic transformation efficiency, rice is also an excellent model system for studying monocotyledonous biology [1]. Since 1997,great efforts have been devoted to deciphering the rice (Oryza sativaL.ssp.japonicaandindica)genomes[2–4],and finally in 2005, representative genomes were assembled into chromosome scale[5,6].It should be noted,however,that any rice genome can be fully utilized only when its high-quality annotation is available; incomplete, incorrect, or ambiguous annotation could bring considerable obstacles for comprehensive characterization of gene function and in-depth exploration of molecular mechanisms underlying complex agronomic traits.Therefore,complete and accurate genome annotation is of fundamental importance in support of yielding scientific findings in rice studies [7–10].
Genome reannotation holds the potential to not only improve structural and functional information but also discover novel protein-coding and non-coding genes. Nowadays,next-generation sequencing(NGS)technologies have triggered an explosion of RNA-seq data,providing great opportunity in genome reannotation.As RNA-seq analysis enables identification of splice junction sites and novel exons with higher confidence [11], there is no doubt that rice genome annotation can be significantly improved based on these precious data, especially when considering the efforts that have already been paid in other species [12–14]. Currently, there are two widely used annotation systems for the rice(O.sativaL.ssp.japonica)genome,namely,MSU-7.0 and RAP-DB[10].However,they were generated mainly based on expressed sequence tags(EST)and cDNA sequences,etc., with limited amount of highthroughput NGS data integrated [10]. Although RNA-seq libraries from various rice tissues and diverse experimental conditions are growing at an unprecedented pace, so far, no attempt has been made to apply all these valuable resources for rice gene model refinement.Therefore,it is highly desirable to reannotate the rice genome based on large-scale integration of high-throughput transcriptomic data.
The Information Commons for Rice(IC4R,http://ic4r.org)[15–17], one of the core resources of National Genomics Data Center(NGDC,http://bigd.big.ac.cn)[18–20],is a public database integrating multiple omics data for rice and providing high-quality annotations. Here, we perform rice genome reannotation based on integration of large-scale RNA-seq data and consequently release a new annotation system—IC4R-2.0 forO. sativaL. ssp. japonica. IC4R-2.0 presents considerable improvements by enhancing structural completeness of protein-coding genes, incorporating an abundance of functional annotations, and systematically identifying long noncoding RNAs (lncRNAs) and circular RNAs (circRNAs) in rice genome. Accordingly, we upgrade the IC4R database by providing more user-friendly web interfaces and implementing a series of practical online tools. Collectively, the improved annotation system IC4R-2.0 as well as the updated database remarkably increase the utility of the rice genome, thereby bearing great promise for comparative and functional genomic studies in rice and other monocotyledonous species.
Method
RNA-seq data collection
More than 1800 RNA-seq datasets ofO.sativaL.ssp.japonicareleased before May 1st, 2017 were downloaded from NCBI Sequence Read Archive (SRA) [21] and NGDC Genome Sequence Archive (GSA) [22]. These datasets were generated from a diversity of rice tissues across various developmental stages and experimental conditions. After removal of libraries with short sequencing reads (average length <36 bp) and unclear meta-information, a total of 1503 RNA-seq datasets(http://ic4r.org/statistics/RNA-Seq-dataset) with approximately 5.32 terabytes in file size (FASTQ format) were used for rice genome reannotation.
Genome reannotation process
RNA-seq datasets in SRA format were converted into FASTQ format by SRA toolkit (v.2.4.2). Raw reads were adaptertrimmed and quality-filtered (Phred score ≥33; read length≥36 bp) using Trimmomatic (v.0.36) [23] with parameters(LEADING: 15, TRAILING: 15, SLIDINGWINDOW:4:15). The reference-based RNA-seq mapping was performed by HISAT aligner (v.2.1.0) [24] with default parameters. Processed reads were aligned against the reference genome (Os-Nipponbare-Reference-IRGSP-1.0), which was obtained from the Rice Genome Annotation Project(http://rice.plantbiology.msu.edu).
The alignment files in SAM format were converted into BAM format and sorted by SAMtools (v.0.1.19) with default parameters. StringTie (v.1.1.2) [24] was then used to assemble the sorted BAM files into transcripts under the guidance of MSU-7.0, and also to estimate their expression levels by transcripts per million (TPM). The junction reads spanning exon–exon sites of transcripts were annotated and calculated using regtools (v.0.5.0). To decrease the noises caused by lowlyexpressed and fragmented sequences,reconstructed transcripts were filtered by a relatively strict threshold (length ≥200 bp;TPM ≥2.0; minimum reads per bp coverage ≥2.5). Meanwhile, exon–exon junction sites of each transcript should be spanned by valid supporting junction reads.After that,GMAP(v.2015)[25]and BLAT(v.35)[26]were used together to alignthe resulting non-redundant transcripts back to their genomic loci and further merge them into more complete and coordinated transcripts. Then, PASA (v.2.1.0) [27] was used to update MSU-7.0 gene models according to the new transcripts.
Regarding lncRNAs, transcripts with single exon, length<200 bp, or ORF size >100 bp were excluded. Afterwards,the remaining transcripts were compared to the updated protein-coding annotation by Cuffcompare (v.2.2.1). Transcripts with relationship ‘u’ (unknown intergenic transcript),‘o’ (generic exonic overlap with a reference), and ‘x’ (natural antisense transcript) were selected for further analysis. All retained transcripts were blasted against the plant protein sequences from UniRef90 [28] using BLASTX (v.2.2.31+)(E-value cutoff: 1E–05) to remove potential protein-coding transcripts. After that, Coding Potential Calculator (CPC)(v.0.9-r2) [29] and LGC (v.1.0) [30] were collectively used for lncRNA identification with default parameters, and only the consensus ones identified by both tools were incorporated into IC4R-2.0.
Among the 1503 RNA-seq datasets, only those generated from RiboMinus or RiboZero sequencing libraries were selected for circRNA identification. After quality control, the remaining clean reads were aligned against the reference genome (Os-Nipponbare-Reference-IRGSP-1.0) by BWA(v.0.7.10-r789). Then, circRNAs were detected and characterized by CIRI (v.2.0.6) [31] through a two-step process: (1)detecting junction reads with paired chiastic clipping (GTAC)signals;(2)detecting additional junction reads and further filtering to remove false positives caused by incorrectly mapped reads.
Characterization of tissue specificity
The expression breadth, coefficient of variance (CV), and tissue specificity index(τ-value)[32]were used to evaluate expression variability for both protein-coding genes and lncRNAs.Specifically,the τ-values vary between 0 and 1,where the lower τ-values represent less variable expression profiles across different tissues,vice versa.A criterion for selection of housekeeping (HK) and tissue-specific (TS) genes was suggested as follows: (1) HK genes are defined as genes with τ-value<0.5 and CV <0.5, and expressed in >80% tissues; (2)TS genes are defined as genes with τ-value ≥ 0.95, and expressed in <15% tissues; (3) expressed invariable genes(EIGs) are defined as a set of strictly defined HK genes with relatively constant expression levels, which have τ-value <0.45 and CV <0.5, and are expressed in >85% tissues [33].
Database implementation
The updated IC4R was implemented by Java Platform Enterprise Edition (J2EE) as the back-end components and deployed in Apache Tomcat Server(an open-source Java Servlet Container) on a CentOS release 6.5 Linux system. Hypertext Markup Language 5 (HTML5), Cascading Style Sheets 3 (CSS3), Asynchronous JavaScript and XML (AJAX),Data-Driven Documents (D3), Bootstrap, and JQuery were used together to provide user-friendly and interactive frontend web interfaces. All annotation data in the updated IC4R were stored and managed in the open-source MySQL relational database system.
Genome reannotation
In this study, we set up the IC4R reannotation pipeline based upon a reference-guided transcript assembly and reconstruction strategy (Figure S1). As a result, a total of 9,826,047 non-repeated transcripts (17.64 GB in FASTA format) are yielded from 1503 public RNA-seq datasets, representing a great abundance of rice transcriptomes from various tissues and diverse experimental conditions. The reconstructed transcripts are mapped back to the reference genome and subsequently merged as coordinated transcripts to further generate the new annotation system—IC4R-2.0.
Structural improvements
In total, IC4R-2.0 comprises 56,221 protein-coding gene loci corresponding to 80,039 mRNAs. Compared to the previous two annotation systems (MSU-7.0 and RAP-DB), the completeness of gene structure is improved in IC4R-2.0, as the mean lengths of mRNAs,coding sequences(CDS),and exons,as well as the average number of exons per transcript, are all increased (Figure 1A–D). Another improvement is an increase in the number of mRNAs attributed to the inclusion of both 5′and 3′untranslated regions (UTRs) (Table 1). Meanwhile, a total of 16.36%rice gene models are identified to possess alternative splicing,corresponding to 1.42 spliced isoforms per gene on average, which is higher than values obtained using previous annotation systems (1.18 for MSU-7.0 and 1.15 for RAP-DB). Apparently, more alternative splicing events with intron retention are identified in IC4R-2.0 (Figure S2).
Based on the large-scale integration of RNA-seq data,more than 27,000 gene loci are improved in IC4R-2.0 with structural modification(including gene extension and gene merging) and novel gene identification. For example, LOC_Os12g32950,previously annotated in MSU-7.0, is extended to be a more complete gene model IC4R-OSJ12G289800 in IC4R-2.0 through adding a 3′boundary exon, which is in fact well supported by RAP-DB (Figure 2A). Another case of gene extension is observed in IC4R-OSJ12G211900, which is improved by not only adjunction of an internal exon to the MSU-7.0 locus, but also inclusion of 5′-UTR and 3′-UTR (Figure 2B).Furthermore, IC4R-2.0 updates 218 loci by gene merging.Specifically, two neighbouring loci (LOC_Os07g47280 and LOC_Os07g47284) that were originally annotated as separate genes in MSU-7.0, are merged together to form a single gene(IC4R-OSJ07G424200) in IC4R-2.0, which consistently contains complete domains of DNA polymerase zeta catalytic(Figure 2C and S3). Strikingly, IC4R-2.0, based on massive RNA-seq data,identifies a total of 456 novel genes,which possess sufficient RNA-seq evidence but have not been reported in any previous annotation systems (Figure 2D). Particularly,these novel loci are further verified via multiple protein sequence alignments;taking IC4R-OSJ01G191000 as an example, its reliability is well supported by protein homologs in other fourOryzaspecies (Figure 3).
Characterization of gene expression patterns
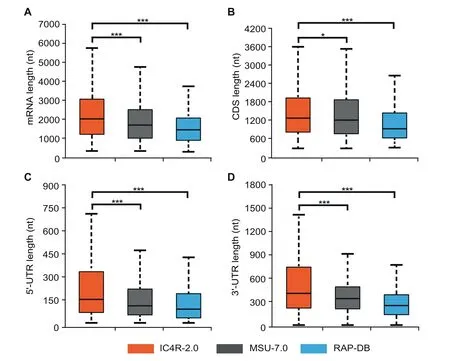
Figure 1 Comparison of structural features among different rice genome annotation systems
To explore the expression patterns of all updated gene models in IC4R-2.0,we investigate their tissue specificity(estimated by τ-value)and expression level(estimated by TPM).When comparing IC4R-2.0 with MSU-7.0 or RAP-DB, all genes can be classified as updated genes (that have different gene structures or are newly identified) and non-updated genes (that have no difference between any two compared annotation systems).Consequently, in contrast to MSU-7.0, we find that the updated gene group in IC4R-2.0 exhibits significantly higher τ-value and lower TPM value than the non-updated gene group. Similar trends are obtained when comparing IC4R-2.0 with RAP-DB (Figure 4A and B). These results clearly demonstrate that genes in the updated group tend to be more tissue-specific and lowly expressed, as RNA-seq data provides higher-resolution transcriptomic evidence and accordingly enable more accurate identification of lowly expressed and/or tissue-specific genes.
Notably,we find that 3996 gene loci annotated in IC4R-2.0 were controversial in the previous annotation systems. For instance, IC4R-OSJ08G082200 and IC4R-OSJ06G014600,were annotated only in one of the annotation systems (RAPDB and MSU-7.0, respectively), without reaching an agreement on gene annotation. Strikingly, IC4R-2.0 verifies the annotation reliability of these two genes with strong evidence from both RNA-seq data (Figure S4A and B) and multiple sequence alignments of the protein products (Figure S5A and B). These results clearly indicate that IC4R-2.0, with the advantage of RNA-seq-based evidence, is capable of bridging the gaps between MSU-7.0 and RAP-DB, and thereby achieves more confident improvements for rice genome annotation.
Functional annotation
IC4R-2.0 additionally presents significant improvements by acquiring multi-level functional annotations. First, all protein sequences identified in IC4R-2.0 are blasted against plant sequences from UniRef90 database. Functional descriptions of the best BLASTP hit (E-value cut-off: 1E–05) are subsequently extracted, corresponding to a total of 47,693 (85.7%)protein-coding genes. Second, a comprehensive set of ontologies, including Gene Ontology (GO), Trait Ontology (TO),Environment Ontology (EO), and Plant Ontology (PO), are retrieved by Blast2GO [34] or via ID mapping to other plant ontology resources. As a result, 43,066 protein-coding genes gain ontology terms. Meanwhile, functional motifs and domains are identified in IC4R-2.0 using InterProScan [35],yielding a total of 155,618 functional entries assigned to 48,159 gene models. Taken together, 55,080 protein-coding genes in IC4R-2.0 are functionally annotated and the detailed statistics are summarized in Table S1.


Figure 2 Structural improvements of IC4R-2.0
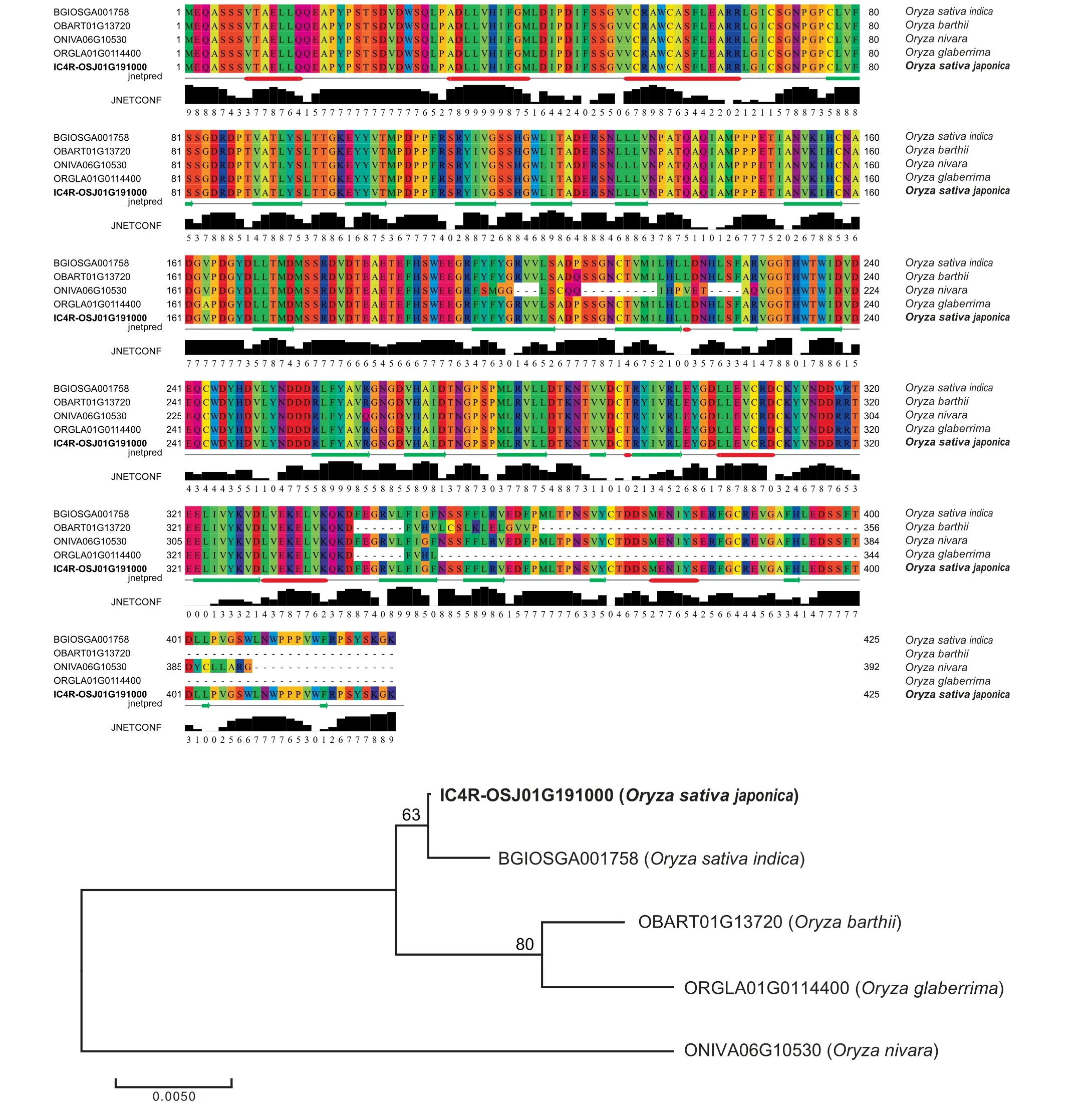
Figure 3 Sequence alignment and phylogenetic tree of a protein encoded by a newly identified gene
Identification of ncRNAs
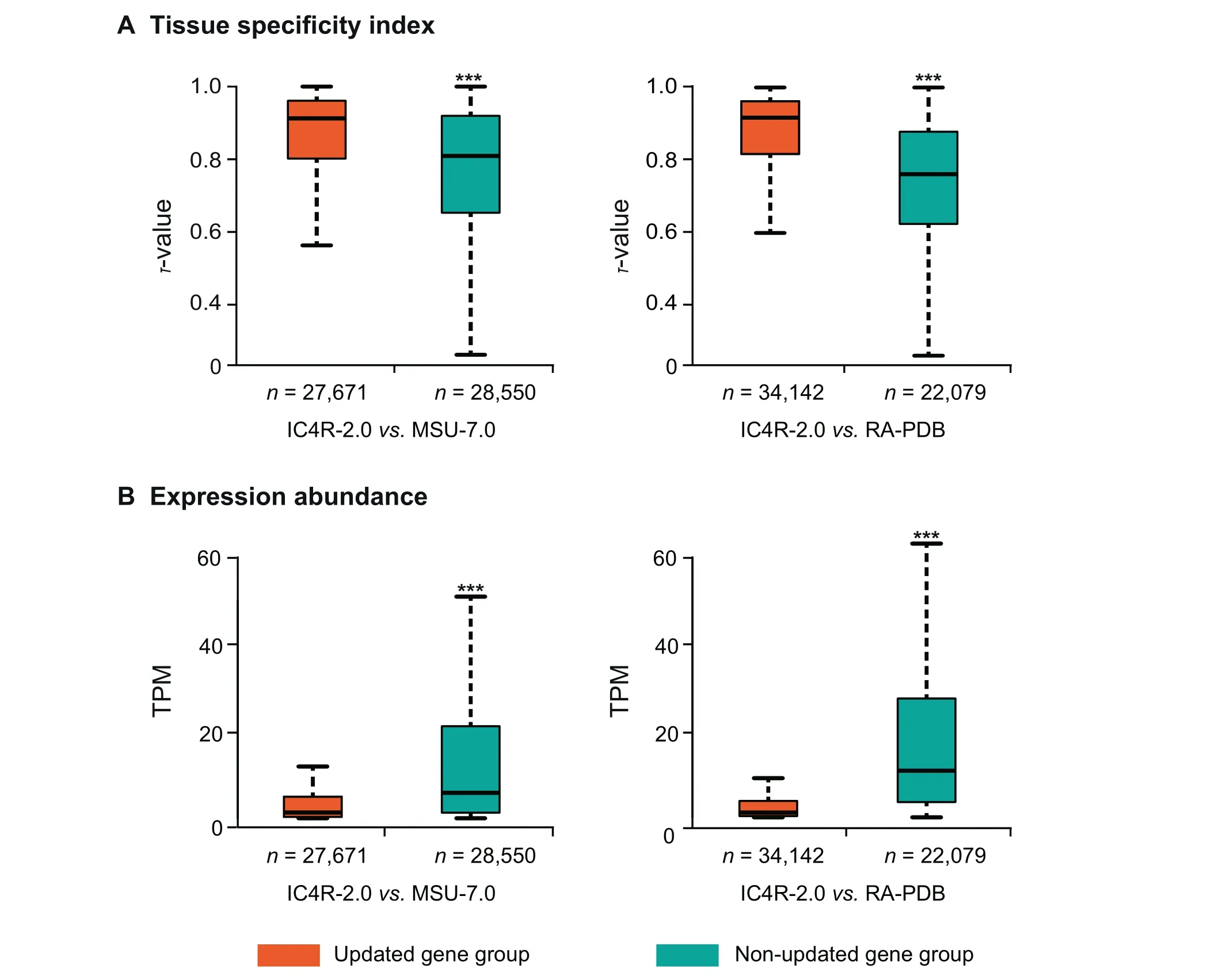
Figure 4 Comparison of tissue specificity index and expression abundance between updated and non-updated gene groups in IC4R-2.0
lncRNAs and circRNAs are important functional regulators involved in many aspects of plant biology [31,36]. Taking advantage of abundant RNA-seq data, we systematically carry out a genome-wide identification of lncRNAs and circRNAs in the rice genome. As a result, 3215 lncRNA loci corresponding to 6259 transcripts are identified (Table 1).Further investigation shows that the size of lncRNA transcripts ranges from 201 to 14,159 bp, with mean length of 1191 bp. Conforming to a previous report [13], lncRNAs,in contrast to protein-coding genes, possess lower expression level and fewer exons(Figure 5A and B).Moreover,narrower expression breadth and higher τ-value are consistently observed in lncRNAs, suggesting that they are likely to be more tissue-specific (Figure 5C and D). Meanwhile, a total of 4373 circRNAs are identified, among which, 3342(76.42%) are exonic, 762 (17.42%) are intergenic, and the remaining 269 (6.16%) are intronic. All these ncRNAs are publicly available at the IC4R website (http://ic4r.org/browse/lncRNA and http://ic4r.org/browse/circRNA).
Evaluation of annotation
We evaluate IC4R-2.0 by comparison with MSU-7.0 and RAP-DB in terms of annotation completeness and quality.First, to assess the completeness of IC4R-2.0, we carry out routine analysis of Benchmarking Universal Single-Copy Orthologs(BUSCO)[37]based on the latest plant dataset(embryophyta odb9). As a result, more complete BUSCO genes are identified in IC4R-2.0 (1389), compared to MSU-7.0(1378) and RAP-DB (1190), and the number of missing BUSCO genes in IC4R-2.0 is reduced accordingly (Table 1 and Figure S6). Additionally, we use MAKER-P software package [38] to evaluate the annotation quality as indicated by Annotation Edit Distance (AED), which ranges from 0 to 1 for each transcript.Specifically,lower AED values represent higher annotation quality,vice versa. Consistently, IC4R-2.0 gives rise to a cumulative curve that shifts to the left, presenting lower AED values than MSU-7.0 or RAP-DB(Figure S7).Together, based on the results shown above, IC4R-2.0 represents a more complete annotation system with better quality,which is primarily attributable to massive RNA-seq data applied in the genome reannotation.
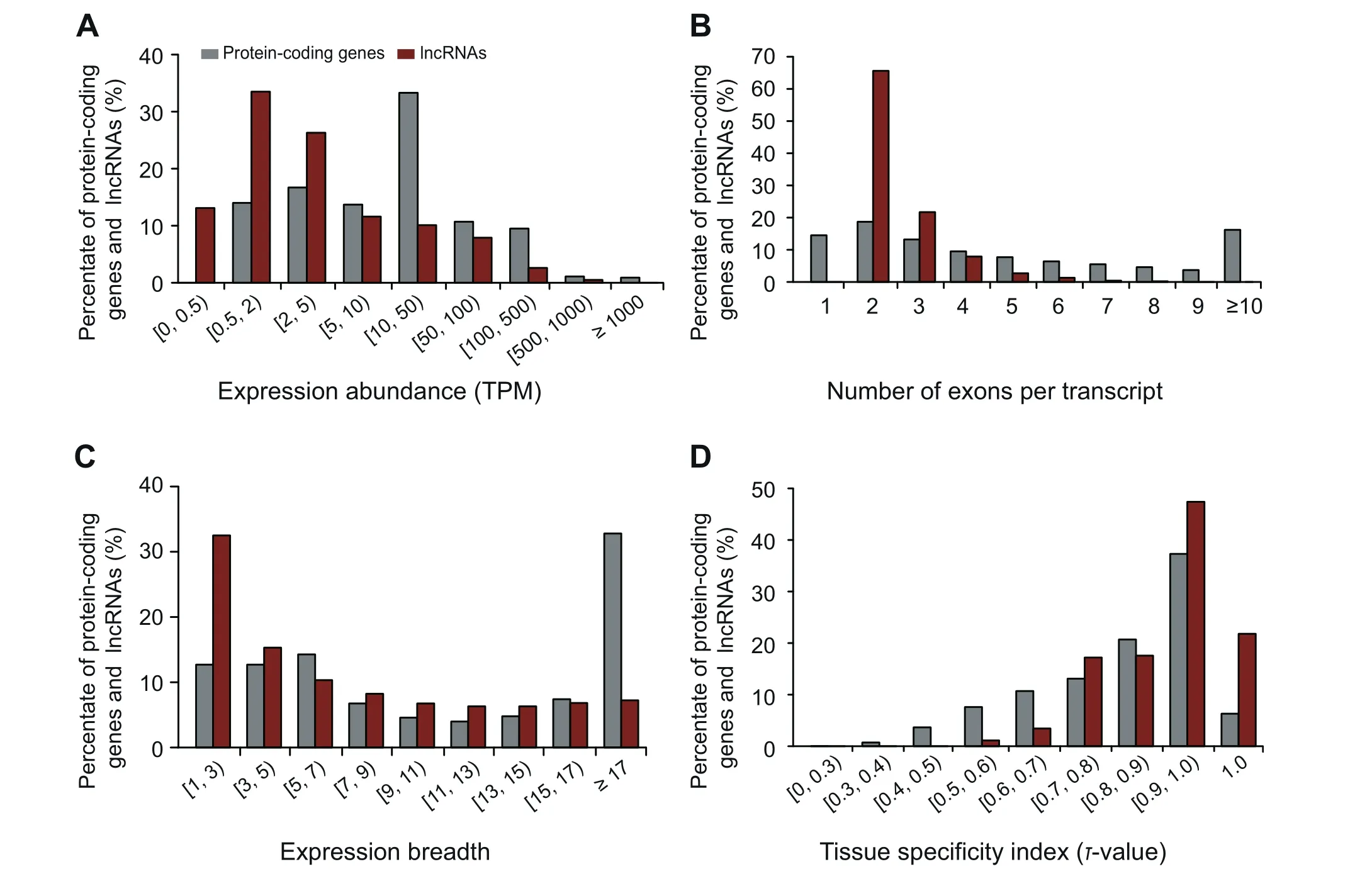
Figure 5 Characterization of lncRNAs in IC4R-2.0
Database update
Data organization and presentation
The IC4R database is significantly updated by incorporating the improved genome annotations and providing userfriendly interfaces for data organization and presentation in light of protein-coding genes, lncRNAs, and circRNAs. For protein-coding genes, basically, IC4R houses a wealth of fundamental information,including gene summary(e.g.,symbols,genomic context, and external hyperlinks), transcripts, associated functional entries, and ontologies (Figure 6A–G). Most importantly, the updated IC4R incorporates abundant information on gene expression, involving expression profiles,expression breadth, τ-value, and associated RNA-seq libraries(Figure 6C). Meanwhile, it features community annotation[39,40], allowing users to contribute their knowledge and expertise to further improvement on gene annotation (Figure 6H). When it comes to lncRNAs, IC4R provides additional information of coding potential scores estimated by both CPC and LGC (Figure S8A–E). Regarding circRNAs,IC4R presents not only basic information, but also Compact Idiosyncratic Gapped Alignment Report (CIGAR) types [31]and the supporting back-spliced junction reads (Figure S9A–D).
Functionality improvement
IC4R is also considerably upgraded by improving multiple functionalities.First,its information retrieval/search functionality is optimized to be more user-friendly and straightforward;it allows a variety of keywords (including gene, lncRNA, circRNA, and domain) as query and also supports fuzzy search.Second, IC4R incorporates a built-in BLAST module and accordingly is capable of sequence similarity search (http://ic4r.org/blast). Third, a web tool named HK-TS Gene Finder(http://ic4r.org/hk-ts)is provided,which is able to identify HK and TS genes with customized criteria. Additionally, a lightweight ID mapping tool(http://ic4r.org/idmapper)is deployed in IC4R, helpful to convert gene IDs among IC4R-2.0, MSU-7.0, and RAP-DB. Last but not least, an interactive genome browser—JBrowse is implemented in IC4R, enabling users to flexibly investigate any given gene, lncRNA, or circRNA in a visualized manner.

Figure 6 Screenshots of protein-coding gene page in IC4R
Enhanced data accessibility
To facilitate access to the new annotation system, IC4R provides a series of flat files for public downloading (http://ic4r.org/download), including gene structural annotation (GFF format), nucleotide and protein sequences (FASTA format),correspondence between IC4R-2.0, MSU-7.0, and RAP-DB ID systems(CSV format),predicted CpG island(TSV format),as well as exon–exon junction information(BED format).Furthermore, to make these associated data accessible more efficiently, an open application programming interface (API)(http://ic4r.org/api) is provided for automatic retrieval.
Conclusions and future directions
Here we have reannotated the rice genome based on integration of large-scale RNA-seq data and accordingly released the new annotation system IC4R-2.0. It significantly updates rice gene models by not only enhancing structural completeness of protein-coding genes, but also identifying novel genes,lncRNAs, and circRNAs. Meanwhile, considerable upgrades are made in IC4R database by implementing more userfriendly interfaces and new functionalities, which together would be of broad utility for functional genomic studies in rice.However,we cannot rule out the possibility that the new annotation system may contain flawed mapping in duplicated genes presumably derived from non-Nipponbare RNA-seq data.Thus, future directions include regular updates of rice reference gene models by integrating more high-quality Nipponbare-specific RNA-seq datasets (especially those with long read length) as well as other types of data (e.g., proteomics data). In addition, more efforts will be devoted to genome-wide reannotation of ncRNAs, such as microRNAs(miRNAs), PIWI-interacting RNAs (piRNAs), small interfering RNAs (siRNAs), and small nucleolar RNAs (snoRNAs).Furthermore, genome reannotation will be conducted for not onlyO. sativaL. ssp. japonica, but also other cultivated and wild rice species.
Availability
The IC4R-2.0 annotation system and related resources are freely accessible at http://ic4r.org/.
Credit author statement
Jian Sang:Investigation,Formal analysis,Software,Visualization, Writing - Original Draft.Dong Zou:Software.Zhennan Wang:Investigation, Formal analysis.Fan Wang:Software.Yuansheng Zhang:Investigation, Data Curation.Lin Xia:Investigation.Zhaohua Li:Investigation.Lina Ma:Formal analysis.Mengwei Li:Formal analysis.Bingxiang Xu:Formal analysis.Xiaonan Liu:Data Curation.Shuangyang Wu:Data Curation.Lin Liu:Data Curation.Guangyi Niu:Data Curation.Man Li:Data Curation.Yingfeng Luo:Data Curation.Songnian Hu:Supervision.Lili Hao:Supervision, Software,Writing -Review &Editing.Zhang Zhang:Supervision, Writing - Review & Editing. All authors read and approved the final manuscript.
Competing interests
The authors have declared that no competing interests exist.
Acknowledgments
This work was supported by grants from the Strategic Priority Research Program of Chinese Academy of Sciences (Grant No.XDA08020102 to ZZ and SH),the Youth Innovation Promotion Association of Chinese Academy of Science (Grant No.2018134 to LH),National Programs for High Technology Research and Development (Grant Nos. 2015AA020108 and 2012AA020409 to ZZ), the 100-Talent Program of Chinese Academy of Sciences (to YB and ZZ), and the National Natural Science Foundation of China (Grant No. 31100915 to LH).
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.gpb.2018.12.011.
ORCID
0000-0001-5393-2520 (Sang, J)
0000-0002-7169-4965 (Zou, D)
0000-0003-4883-2538 (Wang, Z)
0000-0003-0677-6515 (Wang, F)
0000-0001-6876-4611 (Zhang, Y)
0000-0002-4566-9678 (Xia, L)
0000-0002-2673-0103 (Li, Z)
0000-0001-6390-6289 (Ma, L)
0000-0001-6163-2827 (Li, M)
0000-0001-9406-6569 (Xu, B)
0000-0002-0069-1257 (Liu, X)
0000-0001-6305-3204 (Wu, S)
0000-0002-0419-6130 (Liu, L)
0000-0002-8010-8817 (Niu, G)
0000-0002-2799-3431 (Li, M)
0000-0003-1950-9045 (Luo, Y)
0000-0003-3966-3111 (Hu, S)
0000-0003-3432-7151 (Hao, L)
0000-0001-6603-5060 (Zhang, Z)
 Genomics,Proteomics & Bioinformatics2020年2期
Genomics,Proteomics & Bioinformatics2020年2期
- Genomics,Proteomics & Bioinformatics的其它文章
- SuccSite: Incorporating Amino Acid Composition and Informative k-spaced Amino Acid Pairs to Identify Protein Succinylation Sites
- HybridSucc: A Hybrid-learning Architecture for General and Species-specific Succinylation Site Prediction
- BGVD: An Integrated Database for Bovine Sequencing Variations and Selective Signatures
- SR4R: An Integrative SNP Resource for Genomic Breeding and Population Research in Rice
- iGMDR: Integrated Pharmacogenetic Resource Guide to Cancer Therapy and Research
- MosaicBase: A Knowledgebase of Postzygotic Mosaic Variants in Noncancer Disease-related and Healthy Human Individuals