
Oligodendrocyte precursor cell maturation: role of adenosine receptors
2021-01-24 09:15FedericaCherchiAnnaMariaPuglieseElisabettaCoppi
中國(guó)神經(jīng)再生研究(英文版) 2021年9期
Federica Cherchi, Anna Maria Pugliese, Elisabetta Coppi
Abstract Oligodendrocyte-formed myelin sheaths allow fast synaptic transmission in the brain and their degeneration leads to demyelinating diseases such as multiple sclerosis.Remyelination requires the differentiation of oligodendrocyte progenitor cells into mature oligodendrocytes but, in chronic neurodegenerative disorders, remyelination fails due to adverse environment. Therefore, a strategy to prompt oligodendrocyte progenitor cell differentiation towards myelinating oligodendrocytes is required. The neuromodulator adenosine, and its receptors (A1, A2A, A2B and A3 receptors: A1R, A2AR,A2BR and A3R), are crucial mediators in remyelination processes. It is known that A1Rs facilitate oligodendrocyte progenitor cell maturation and migration whereas the A3Rs initiates apoptosis in oligodendrocyte progenitor cells. Our group of research contributed to the field by demonstrating that A2AR and A2BR inhibit oligodendrocyte progenitor cell maturation by reducing voltage-dependent K+ currents necessary for cell differentiation.The present review summarizes the possible role of adenosine receptor ligands as potential therapeutic targets in demyelinating pathologies such as multiple sclerosis.
Key Words: adenosine receptors; K+ channels; oligodendrocyte differentiation;oligodendrocyte progenitor cells; remyelination
Introduction
Oligodendrocytes (OLs) are glial cells in the central nervous system (CNS), responsible for myelin sheath formation, which allows fast signal transmission and provides metabolic support to axons (Saab et al., 2016; Simons and Nave, 2016).
During development, OLs are generated in the germinal zone from migratory bipolar oligodendrocyte precursor cells (OPCs),which are renowned for the expression of the proteoglycan nerve-glial antigen 2 (NG2) (Sakry and Trotter, 2016).
Most OPCs differentiate into myelinating OLs, however a pool of immature OPCs, comprising the 5-8% of total glial cells, persists within the adult CNS where they represent the major population of proliferating cells (Dawson et al., 2003;Domingues et al., 2018) and retain the ability to generate new OLs. This process guarantees, under physiological conditions,myelin turnover and remodeling in response to life experience(Bergles and Richardson, 2016) and, in conditions of tissue damage, the generation of new myelin, an ability that might be lost through normal aging or chronic diseases.
Recruitment of OPCs to the lesions is in fact the most important event for remyelination after CNS injury or in demyelinating diseases such as multiple sclerosis (MS)(Neumann et al., 2019). Unfortunately, for unknown reasons,this process sometimes fails, or is insufficient to provide myelin repair, in particular during chronicization of the disease. Thus,increasing interest is focusing on OPCs as therapeutic target in demyelinating disorders, since fostering OPC maturation into myelinating OLs could prevent disease progression and the consequent disability (Skaper et al., 2019).
Search Strategy and Selection Criteria
This review was compiled by using “PubMed” with sources within the last 5 years, with an emphasis on the most recent,novel, and comprehensive papers. If the topic did not have relevant information within the last 5 years, we used the most recent paper. Due to the strict limit of 50-100 references, we could not cite all of the relevant publications.
Oligodendrocyte Differentiation
Before being able to produce myelin, oligodendroglial cells progress through a series of highly regulated steps of differentiation from OPCs to mature OLs (Kuhn et al., 2019).During embryonic development, OPCs are generated in restricted areas, such as the subventricular zone, and present a significant migratory ability that allow them to spread and populate the brain and spinal cord (Kuhn et al., 2019).Their differentiation and maturation are postnatal processes characterized by the loss of proliferative activity and the acquisition of an elaborate morphology with highly branched processes (Kuhn et al., 2019). Oligodendrogliogenesis involves a sequence of distinct phases that can be identified by the expression of stage-specific surface antigens and by morphological changes (Kuhn et al., 2019). On these bases, a classification into three stages of differentiation has been proposed: proliferating OPCs, post-mitotic preoligodendrocytes (pre-OLs) and mature myelinating oligodendrocytes (OLs) (Coppi et al., 2013, 2015; Kuhn et al.,2019). The initial stage of maturation presents a bipolar (or tripolar) morphology, typical of proliferating OPCs (Fumagalli et al., 2011). Several are the markers of precocious maturation stages, such as platelet-derived growth factor (PDGF) receptor alpha (PDGFα), NG2 or the transcription factor Olig2 (Pringle et al., 1992; Sakry and Trotter, 2016).
When OPCs start to differentiate in pre-OLs, secondary ramifications emerge from the soma and the expression of new molecular markers, typical of intermediate steps of maturation, is detected, such as O4 (Kuhn et al., 2019) and the recently deorphanized P2Y-like GPR17 receptor (Lecca et al., 2008; Fumagalli et al., 2011; Kuhn et al., 2019). During this phase, cells acquire the typical phenotype of postmitotic, but not yet myelinating, immature OLs characterized by a complex multipolar morphology (Kuhn et al., 2019).
Finally, when OLs reach the fully mature, myelinating phase,they acquire a highly ramified profile and immunoreactivity for myelin specific structural proteins such as 2′,3′-cyclicnucleotide-3′-phosphodiesterase, myelin associated glycoprotein (MAG) and myelin basic protein (MBP) (Kuhn et al., 2019). Mature OLs synthesize large amounts of myelin,giving rise to multilamellar myelin sheaths that wrap and insulate neuronal axons, which allow electrical isolation and saltatory conduction of electric impulses.
It is known that, during their maturation, oligodendroglial cells display different functional voltage-gated ion channels (Sontheimer and Kettenmann, 1988; Barres et al., 1990; Berret et al., 2017; Spitzer et al., 2019), including either inward or outward rectifying K+channels, Na+currents and different subtypes of Ca2+channels (Verkhratsky et al., 1990) and the density of channels differs within age and region (Spitzer et al., 2019). Such a heterogeneity may therefore reflect different cellular states, where densities of ion channels define a particular cell function.
When OPCs first appear, i.e. at embryonic day 13 (E13)in the mouse, they have no detectable voltage-gated ion channels nor glutamate receptors and may be therefore considered in a naive state (Spitzer et al., 2019). The first ion channels detected are KVand α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid and kainate receptors (AMPARs/KARs), at E18. OPCs with these properties are considered migrating cells because of the strong expression of migratory genes at this time point. The fraction of OPCs with detectable NaVincreases sharply around birth. It is conceivable that OPCs with high NaVand KV, and low AMPA/KAR, densities reflect a high proliferation state because (1) OPCs in S/G2/M phase have a higher density of NaVthan OPCs in G0/G1phase (Spitzer et al., 2019), (2) the higher the fraction of this type of OPC,the higher the proportion of OPCs in G2/M phase (Spitzer et al., 2019), (3) proliferating 5-ethynyl-2-deoxyuridine (EdU)-positive OPCs show this pattern of ion channel expression(Clarke et al., 2012), (4) this state of OPCs is the most prominent during the OPC recruitment phase (the period of highest proliferation) in myelin regeneration (Gautier et al.,2015). OPCs expressing NaV, KV, AMPA/KARs, and N-methyl-Daspartate receptors (NMDARs) are typically found throughout oligodendrogliogenesis during development, when myelin gene expression starts (Marques et al., 2018; Spitzer et al.,2019), and during the beginning of the differentiation phase of myelin regeneration (Gautier et al., 2015) and so it might reflect a ‘‘primed’’ OPC state for differentiation. Either KVor AMPA/KAR channels were expressed in nearly all recorded postnatal OPCs, whereas, intriguingly, not all OPCs express NaVor NMDARs, as their density reaches a maximum after the first postnatal week, when myelination starts, and then declines when myelination decays. The last state of OPC maturation is distinguished by low NaVdensity, lack of NMDARs and high AMPA/KAR density and is observed at a time when OPC cellcycle time lengthens, differentiation genes are downregulated and senescent molecular signature genes appear. In this phase, OPCs differentiation potential declines and thus it can be considered a “quiescent” OPC state.
Among K+currents, OPCs show outward currents conductances mainly composed by delayed rectifying K+currents (IK) (Sontheimer and Kettenmann, 1988) characterized by scarce time- and voltage-dependent inactivation and by a threshold for activation around -40 mV. They also express a transient outward K+current, orIA, which is typically found in undifferentiated OPCs and presents a rapid time-dependent inactivation (approximately 50 ms) and a voltage-dependent inactivation at potentials from -40 and above (Gallo et al.,1996). A subpopulation (about 60%) of immature OPCs also express inward, tetrodotoxin-sensitive, Na+currents (INa)typically found in neurons, with a rapid time-dependent inactivation (less than 1 ms) and a current peak amplitude at about -10 mV (Kettenmann et al., 1991).INais never observed in mature oligodendroglial stages, as previously reported by us (Coppi et al., 2013) and others (Sontheimer et al., 1989). Of note, a subpopulation of electrically excitable, spiking, NG2+OPCs, able to generate full action potentials when stimulated by depolarizing current injection, have been described in brain slices, but the functional role of this “electrically excitable”O(jiān)PC subpopulation is still unknown (Káradóttir et al., 2008).Of note, single action potentials have also been detected in a minority of cultured OPCs (Barres et al., 1990).
During maturation, membrane outward K+conductances(bothIKandIA) in OPC undergo a strong downregulation up to almost completely disappearance in mature OLs (Sontheimer and Kettenmann, 1988; Barres et al., 1990; Coppi et al.,2013). In parallel with outward K+current downregulation,there is a gradual increase in the expression of inwardly rectifying K+currents (Kir), activated at potentials lower than-100 mV. Indeed, Kir currents are the main conductance observed in mature OLs (Knutson et al., 1997; Papanikolaou et al., 2020). Among the mentioned currents,IKare crucially linked to cell cycle regulation and hence to myelin formation(Chittajallu et al., 2005) because of the following: (1) a downregulation ofIKoccurs as oligodendrocyte lineage cells mature (Sontheimer and Kettenmann, 1988; Barres et al.,1990) and (2) pharmacological block ofIKinduced by tetraethyl-ammonium in cultured OPCs is sufficient to inhibit their proliferation and differentiation (Gallo et al., 1996; Knutson et al., 1997; Chittajallu et al., 2005; Coppi et al., 2013). Hence,treatments aimed at modulating these currents may affect oligodendrocyte proliferation and myelination.
Steps and markers of oligondendroglial differentiation described above are observed not only in the brain but also in the spinal cord, where a significant fraction of OPCs also persists throughout adult life.
What is clear is that these changes in voltage-gated channels will have a profound effect on how OPCs sense neuronal activity and on the effect neuronal inputs will have on OPCs(Coppi et al., 2013; Spitzer et al., 2019).
Neurotransmitters, cytokines and growth factors have been shown to regulate glutamate receptor expression in OPCs(Stellwagen and Malenka, 2006; Malerba et al., 2015; Spitzer et al., 2019). Accordingly, a combination of G-protein coupled receptors, growth factors, and cytokines may modify K+current expression. This heterogeneity in physiological properties may cause differences in the myelination potential of OPCs and implicate distinct functions or cell states (Figure 1).
Adenosine and Oligodendrocyte Maturation
Adenosine is an endogenous neuromodulator that recently emerged as an important mechanism for intercellular communication in the nervous system (Pucha?owicz et al., 2015). Its actions are mediated by the activation of metabotropic P1 purinergic receptors: in general, A1and A3receptors (A1R, A3R) are coupled with Gi, Gqand Goprotein,while A2Aand A2Breceptors (A2AR, A2BR) relies on the stimulation of adenylyl cyclase (AC) by Gsor Golf(Antonioli et al., 2013). In addition to Gs, A2BR receptors can activate phospholipase C through Gq(Antonioli et al., 2013).
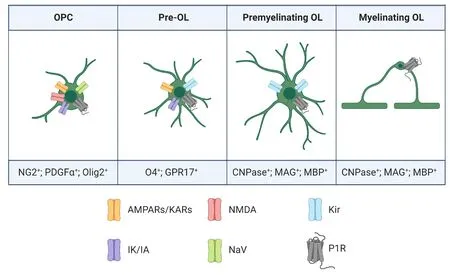
Figure 1|Schematic representation of morphological and antigen/channel expression changes during oligodendrogliogenesis.A typical oligodendrocyte precursor cell (OPC) is positive to the antigens:nerve glial antigen 2 (NG2+), platelet-derived growth factor alpha (PDGFα+)and to the transcription factor Olig2 (Olig2+) and express glutamate AMPA and/or kainate receptors (AMPARs/KARs) and voltage-dependent Na+ (Nav)and K+ (IK/IA) channels. A typical pre-oligodendrocyte (Pre-OL) is positive to the markers: oligodendrocyte 4 (O4+), the purinergic-like receptor GPR17(GPR17+) and express AMPARs/KARs, inward-rectifier potassium channels (Kir)and IK/IA channels. Premyelinating OLs and myelinating OLs are positive to the antigens: 2′,3′-Cyclic-nucleotide-3′-phosphodiesterase (CNPase+), myelin associated glycoprotein (MAG+) and myelin basic protein (MBP+) and express Kir channels. During oligodendrogenesis P1Rs are expressed at all maturation stages.
All P1 receptors are expressed at all maturational stages of oligodendroglial cells (Stevens et al., 2002; Fields, 2004)and exert a key role in cell development. Furthermore, the expression by oligodendrocytes of the equilibrative nucleoside transporters ENT1 and ENT2, as well as adenosine degrading enzymes, such as adenosine deaminase and adenosine kinase,suggests that these cells are able to sense and finely tune extracellular adenosine levels (González-Fernández et al.,2014), thus supporting the notion that purinergic signaling exerts a prominent role in these cells (Burnstock et al., 2011).Indeed, our group of research contributed to demonstrate that adenosine can affect numerous OPC functions such as migration, proliferation and maturation (Stevens et al., 2002;Fields, 2004; Coppi et al., 2013, 2015), with distinct effects mediated by different receptor subtypes, as described below.
A1R in oligodendrogenesis
Exogenous adenosine added to OPCs cultured in the presence of the mitogen PDGF, leads to a concentration-dependent reduction of cell proliferation and promotes differentiation towards pre-myelinating oligodendrocytes, an effect that is mainly mediated by A1Rs (Stevens et al., 2002). Furthermore,tonic electrical stimulation of co-cultures of OPCs with dorsal root ganglion neurons also promotes myelination by increasing the number of MBP+cells (Stevens et al., 2002), an effect blocked by a cocktail of A1R, A2AR and A3R antagonists,suggesting that endogenous adenosine released in response to impulse activity promotes oligodendrocyte development and myelination (Stevens et al., 2002). In addition, A1R agonists have been reported to stimulate OPC migration(Othman et al., 2003). On these bases, it was proposed that A1Rs on OPCs prompt myelination thus offering new approaches for the treatment of demyelinating diseases of the CNS, such as MS. In accordance, A1R-/-mice developed more severe experimental autoimmune encephalomyelitis(EAE; a mouse model of MS) with worsened demyelination,axonal injury, and enhanced neuroinflammation and activation of microglia/macrophages (Tsutsui et al., 2004). Furthermore,A1Rs promotes myelin repair by recruiting endogenous progenitor cells in an experimental model of optic nerve demyelination (Asghari et al., 2013) and, when activated on astrocytes, exert immunosuppressive properties (Liu et al.,2018a).
Such protective effects, however, are at variance from what has been described inin vivoneonatal rats, where the treatment with A1R agonists reduces white and gray matter volume, induces ventriculomegaly (Turner et al.,2003) and decreases the expression of MBP, similarly to what observed in neonatal rats reared in hypoxia (Ment et al., 1998). Ventriculomegaly was also observed in mice lacking the enzyme adenosine deaminase which degrades adenosine (Turner et al., 2003). Moreover, hypoxia-induced periventricular white matter injury (PWMI, a form of brain injury present in preterm infants) was prevented in A1R-/-mice (Turner et al., 2003). These data support the notion that adenosine, acting on A1Rs, mediates hypoxia-induced brain injury and ventriculomegaly during early postnatal development. Such an effect could be attributed to the fact that adenosine, which is released in huge amounts during hypoxic-ischemic conditions (Latini and Pedata, 2001),activates A1Rs leading to premature differentiation and reduced proliferation of oligodendroglia precursors. Indeed,studies on OPCs and pre-OLs in hypoxic conditions, when increased glutamate outflow impairs neuronal functions(Rossi et al., 2000) and synaptic transmission (Colotta et al.,2012), revealed a reduced proliferation and an accelerated maturation, as demonstrated by the increased expression of the cell cycle regulatory proteins p27 (Kip1) and phosphocdc2 (Akundi and Rivkees, 2009). This series of events would lead to a reduced number of OLs available for myelination,thus contributing to PWMI (Rivkees and Wendler, 2011). So,strategies aimed at stimulating OPC proliferation in neonatal hypoxia/ischemia may be of value to prevent PWMI.
Accordingly, Cao and co-workers (Cao et al., 2019) found that OLs pre-treated with 100 μM caffeine or the A1antagonist 8-cyclopentyl-1,3-dipropylxanthine (DPCPX; 100 nM) during hypoxia showed a significant reduction in A1R and Olig2 expression, at early stages, and a decreased 2′,3′-Cyclicnucleotide-3′-phosphodiesterase expression, at later stages of hypoxia. In addition, they demonstrated that either hypoxia or adenosine treatment induced significant elevation in resting[Ca2+]i, which was restored to normal levels when cells were treated with caffeine or DPCPX. During hypoxia, adenosine increase leads to A1R activation which resulted in excessive Ca2+release from intracellular stores (Annunziato et al., 2013),a condition that is considered to initiate cell injury (Cao et al.,2019).
A2AR in oligodendrogenesis
The first functional characterization of the adenosine A2AR subtype in OPCs has been reported by our group of research(Coppi et al., 2013). We demonstrated that the selective A2AR agonist CGS21680 inhibitsIKcurrents in cultured OPCs and delaysin vitroOPC differentiation since it increases the percentage of NG2+immature OPCs and reduces O4+pre-OLs and MAG+mature OLs along 12 days of cell culture,without affecting neither cell viability nor proliferation (Coppi et al., 2013). These effects were completely prevented in the presence of the selective A2AR antagonist SCH58261 (Coppi et al., 2013). Tetra-ethyl-ammonium, at 3 mM concentration which blocks sustainedIKbut not transientIAcurrents in cultured OPCs, mimics and occludes the effect of the A2AR agonist on membrane currents, confirming that this purinergic receptor subtype electively affectsIKin cultured OPC (Coppi et al., 2013). In keeping with data demonstrating thatIKinhibition impairs proliferation and maturation of cultured OPCs (Gallo et al., 1996; Coppi et al., 2013) and blocks myelin deposition in the embryonic spinal cord (Shrager and Novakovic, 1995),it appears that A2AR stimulation inhibits OPC differentiation by reducingIKcurrents. In line with this assumption is the observation that selective activation GPR17, a Gi-coupled P2Ylike receptor, enhances tetra-ethyl-ammonium-sensitiveIKand improves OPC differentiation (Coppi et al., 2015).
Recently, Fontenas and colleagues (Fontenas et al., 2019)demonstrated that the A2AR antagonist SCH-58261 induced ectopic OPC migration from motor exit point in transition zones in zebrafish larvae, an effect that is not shared by antagonists at the other adenosine receptor subtype .
A pathological condition associated with defects in cell metabolism and OPC maturation is the Niemann-Pick type C 1 (NPC) disease, an autosomal recessive and progressive neurovisceral disorder characterized by intracellular cholesterol accumulation and myelin defects (Kodachi et al.,2017). De Nuccio and colleagues observed that in primary cultures of OPCs exposed to a cholesterol transport inhibitor(U18666a), used to induce the NPC1-like phenotypein vitro, A2AR expression was significantly decreased whereas treatment with the A2AR agonist CGS21680 triggered a protective effect by reducing cholesterol accumulation and mitochondrial membrane potential (mMP) alterations in U18666a-treated OPCs (De Nuccio et al., 2019). Consistent with data from Coppi and colleagues, the same study demonstrates that CGS21680 induced a decrease in the percentage of O4+, O1+and MBP+in control OPCs (Coppi et al., 2013; De Nuccio et al., 2019). In contrast, after 48 hours of U18666a treatment, CGS21680 overcame the maturation arrest induced by the compound, even when A2AR stimulation occurred 24 hours after U18666a exposure. Finally, the same study also demonstrated that protein-kinase A (PKA) activation is responsible for the A2AR-dependent effect on cholesterol accumulation since the PKA inhibitor KT5720, but not the extracellular signal-regulated kinases 1/2 (ERK1/2) inhibitor PD98059, prevented the cholesterol redistribution induced by CGS21680 in NPC-OPC. The dual effect of CGS21680 on OPC differentiation, arresting OLs maturation in control cultures and promoting differentiation in U18666a-treated cultures, is in keeping with differential effects by CGS21680 previously reported in a model of Huntington’s disease, where the compound induces opposite effects in the striatum of Huntington versus wild-type mice (Martire et al., 2007).
However, other intracellular pathways, in addition toIKblock, could contribute to the A2AR-mediated inhibition of OPC differentiation. OPCs also express the tyrosine kinase fibroblast growth factor (FGF) receptor whose activation promotes cell proliferation and inhibits the expression of myelin components (Besnard et al., 1989). As an example, in PC12 cells (a cell line that was confirmed to express the A2AR and FGFRs), the simultaneous activation of both A2AR and FGF receptors by robust activation of the mitogen activated protein kinase (MAPK/ERK) pathway, brings to increased differentiation and neurite extension (Flajolet et al., 2008). It is possible that a cross-talk between A2ARs and FGF receptors regulates cell maturation also in OPCs.
Of note, upregulation of A2AR expression has been observed in cerebral white matter of patients with secondary progressive MS and a higher density of brain A2AR appeared to correlate with higher disability scale scores in MS patients (Rissanen et al., 2013). On these bases, it has been hypothesized that A2AR upregulation on brain cells is associated with disease progression. In agreement, in EAE, A2AR antagonists protected from disease development (Mills et al., 2012), suggesting that activation of A2ARs glial and neuronal cells is responsible for EAE development in mice. Moreover, in a rat model of focal brain ischemia (by middle cerebral artery occlusion:MCAo), the myelin damage inflicted to the striatum by the ischemic insult is significantly prevented by the A2AR antagonist SCH58261 that reduced the activation of JNK mitogen activated kinase in oligodendrocytes and subsequent activation of caspase3-mediated oligodendrocyte cell death(Melani et al., 2009).
In keeping with these data, it can be concluded that the activation of A2ARs by adenosine released during a demyelinating insult contributes to brain damage by hampering OPC maturation and myelin deposition. Such a role might appear in contrast with the observation that A2AR agonists proved protective in EAE models by decreasing immune cell infiltration and lymphocyte Th1 cell activation(Liu et al., 2018b). Furthermore, genetic ablation of both central and peripheral A2ARs exacerbates brain damage and neuroinflammation in EAE (Ingwersen et al., 2016). Indeed,A2ARs expressed on peripheral leucocytes are known to exert important anti-inflammatory effects, i.e. by reducing adhesion cell factor production and neutrophil activation (Sitkovsky et al., 2004). Thus, genetic ablation of adenosine A2ARs on blood cells exacerbates leucocyte infiltration, neuroinflammation and brain damage in a model of chronic inflammation such as EAE(Pedata et al., 2014). It appears that, beside disadvantageous central effects on OPC differentiation, A2AR stimulation may also alleviate neuroinflammation by peripheral mechanisms,thus complicating the role of this endogenous nucleoside in neurodegenerative diseases. Successive studies contributed to elucidate the multifaceted role played by A2ARs in EAE.Ingwersen and colleagues demonstrated that A2ARs were upregulated predominantly on T cells and macrophages/microglia within the inflamed tissue and preventive EAE treatment with A2AR-specific agonist inhibited myelinspecific T cell proliferationex vivoand ameliorated disease,while application of the same agonist after disease onset exacerbated non-remitting EAE progression and tissue damage(Ingwersen et al., 2016). Similarly, Chen and co-workers(Chen et al., 2019) demonstrated that the administration of the selective A2AR antagonist SCH58261 at 11-28 days postimmunization with MOG prevented neurological deficits and reduced local infiltration and demyelination. By contrast, the same treatment was ineffective when administered at the beginning of the onset of EAE (i.e., 1-10 after immunization).Therefore, it appears that, while providing anti-inflammatory effects on T cells and thus protection at early stages, A2AR seems to play a detrimental role during later stages of the disease and may thus contribute to sustained tissue damage within the inflamed CNS. Hence, the identification of the effective therapeutic window to optimize the beneficial effects of A2AR antagonists is of crucial importance to support SCH58261 as a candidate for the treatment of MS in human(Rajasundaram, 2018).
A2BR in oligodendrogenesis
We previously demonstrated that also A2BRs are crucially involved in OPC maturation. Similarly to what observed in the presence of the A2AR agonist CGS21680 (Coppi et al., 2013),we found that the selective A2BR agonist BAY60-6583 (10 μM)reversibly inhibits outwardIKand alsoIAconductances, as opposed to A2AR stimulation which only inhibitIK, in cultured OPCs (Coppi et al., 2020). Similar results were found in the presence of the recently synthetized and selective A2BR agonist P453 (50 nM), described by Betti and co-workers(Betti et al., 2018), and the unselective adenosine receptor agonist NECA (50 μM) applied in the continuous presence of saturating concentrations of A1R, A2AR and A3R antagonists(DPCPX, SCH58261 and MRS1523, respectively; all 100 nM).Similar K+current inhibition in the presence of BAY60-6583 was obtained in mature OLs. In addition, we found that the AC activator forskolin also inhibited K+currents and occluded the effect of a further application of BAY60-6583, suggesting that the A2BR-mediated effect involves intracellular [cAMP]rise. Silencing experiments in the same work indicated that A2BR downregulation by small interference RNA (siRNA) is,рer se, a signal to enhance OPC differentiation. Given the notoriously low affinity of this receptor subtype for adenosine,we discourage the hypothesis of A2BR being activated by endogenous adenosine released by OPCs or of constitutive A2BR activation, because we did not observe any modification in K+currents when the A2BR antagonist MRS1706 was applied alone. For these reasons, in trying to provide an explanation for the effect of siRNA-A2BRрer sein OPC cultures, we hypothesize that A2BR could possibly dimerize with some other receptors (an option that has been previously reported for this adenosine receptor subtype; Fusco et al., 2018, 2019). Our data are in line with those reported by Wei and co-workers,who demonstrate that pharmacological blockade of A2BR with selective antagonists or receptor knock out in a rodent model of EAE, protects from myelin disruption and neurological impairment due to this pathological condition (Wei et al.,2013). However, unless the lack of preclinical studies where A2BR agonists are administered in EAE mice up to date, it cannot be ignored that the A2BR subtype shares with the A2AR the anti-inflammatory impact in many different pathologies(Pedata et al., 2016; Dettori et al., 2020) indeed, eventual side effects could arise in MS patients treated with A2BR blockers.
A3R in OL survival
No data are present in the literature about the effect/s of A3Rs on oligodendrocyte differentiation. However, results obtained by Gonzalez-Fernandez and colleagues (González-Fernández et al., 2014) demonstrate that the A3R agonist 2-CI-IB-MECA induces apoptosis of cultured O4+oligodendrocytes isolated from rat optic nerve through the activation of Bcl-2-associated X (Bax) and p53-upregulated modulator of apoptosis (PUMA)proapoptotic proteins. Furthermore, incubation ofex vivopreparations of optic nerve with adenosine or 2-CI-IB-MECA induces OL damage and myelin loss, effects prevented by the A3R antagonist MRS220 (González-Fernández et al., 2014).Moreover MRS220 also prevented oligodendrocyte damage and myelin loss in the optic nerve exposed toin vitroischemic like conditions, i.e. oxygen-glucose deprivation (González-Fernández et al., 2014). Thus, data suggest that adenosine,via activation of A3Rs, triggers oligodendrocyte death and contributes to white matter ischemic damage.
Concluding Remarks
As widely described above, one of the pervasive mechanisms by which released adenosine affects OPC functionality is ion channel regulation. This notion is in line with previous findings demonstrating that voltage-dependent channels can regulate many signaling pathways involved in proliferation by modulating the electrical, mechanical and chemical properties of cells (Rao et al., 2015).
Accordingly, in OPC cultures, a link between K+currents,proliferation, differentiation and myelination has been demonstrated (Chittajallu et al., 2002), i.e. Kvchannels regulate membrane potential and cell volume, and subsequently could modulate cell cycle progression (Pérez-García et al., 2018).
Above data demonstrate that the Gs-coupled A2AR and Gs/qcoupled A2BR subtypes decreaseIK. K+channel modulation by adenosine receptors has already been described in other cell types (Xu and Enyeart, 1999), with an involvement of either intracellular cAMP rise or a direct action of the G protein coupled to receptors being hypothesized. The relationship between cAMP andIKis supported by our recent experimental evidence that exogenous application of forskolin, a direct AC activator, is sufficient to decreaseIKcurrents in cultured OPCs (Figure 2) (Coppi et al., 2020). Interestingly, previous observations have shown that forskolin inhibits alsoin vitromyelin deposition by OPCs (Vartanian et al., 1986). Of note,activation of GPR17, a deorphanized Gi-coupled P2Y-like receptor, which stimulatesin vivo(Parravicini et al., 2020)andin vitroOPC differentiation (Lecca et al., 2008; Fumagalli et al., 2011), elicits opposite effects in comparison to the Gscoupled A2AR and A2BR subtypes, i.e. it increasesIKin cultured OPCs (Coppi et al., 2013). Taken together, data suggest that intracellular signaling pathways leading to cAMP decrease in OPC cultures are coupled toIKstimulation and increased cell differentiation whereas those mediating intracellular cAMP rise inhibitIKand also OPC differentiation (Figure 1). Such a relationship agrees with previous observations made on ovine OLs (Soliven et al., 1988).
As evaluated by studies on cultured OPCs, adenosine acts as a dual modulator of OPC development. By stimulating A1Rs, which increaseIK, adenosine promotes oligodendrocyte proliferation and maturation. On the contrary, by stimulating A2ARs or A2BRs, which decreaseIK, adenosine inhibits oligodendrocyte differentiation. The A3R subtype is an inducer of OL apoptosis. Overall, the outcome of adenosine-mediated effects on myelin deposition likely depends on the timing of receptor stimulation during OPC development or in distinct phases of demyelinating pathologies. For example, the fact that OPCs are present in MS lesions but fail to differentiate into mature OLs (Dulamea, 2017) suggests that remyelination processes are blocked at a pre-myelinating stage. Due to the important actions of adenosine onIKcurrents and, in turn,on OPC maturation (Table 1), brain A1Rs, A2ARs and A2BRs might represent new molecular targets to develop innovative pharmacological tools in demyelinating pathologies, such as MS, stroke and brain trauma.
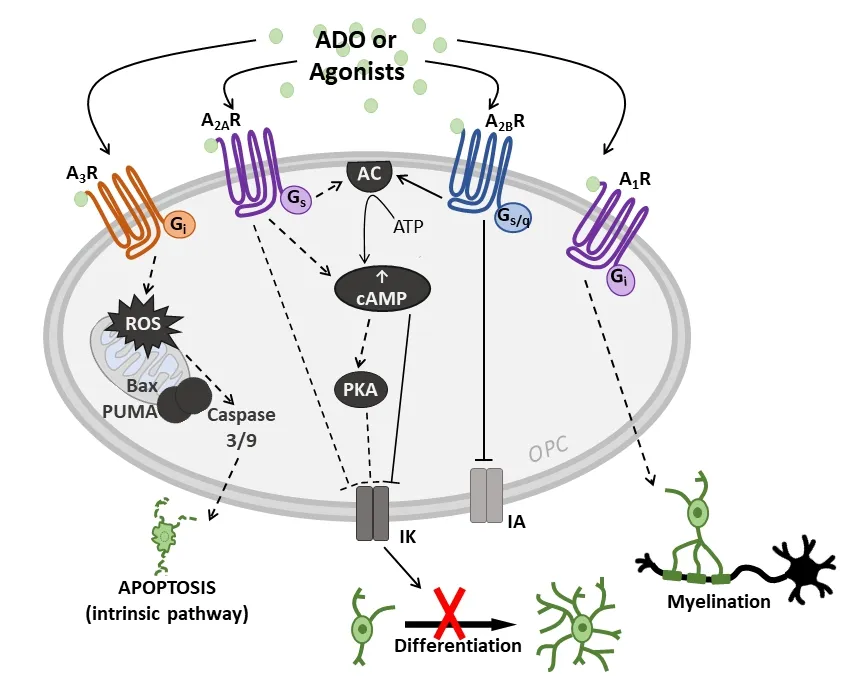
Figure 2|Effects of A1, A2A, A2B and A3 receptor (A1R, A2AR, A2BR and A3R)activation on oligodendrocyte precursor cells (OPCs) and intracellular pathways involved.The activation of A1Rs by adenosine (ADO) or other receptor agonists facilitates myelin deposition by Gi coupling. The stimulation of Gs-coupled receptors A2AR and/or A2BR lead to adenylyl cyclase (AC) activation causing an increase in intracellular cyclic adenosine monophosphate (cAMP),which, in turn, closes IK channels and inhibits OPC differentiation possibly by a mechanism involving protein kinase A (PKA) activation. A2BR activation also induces a block in IA currents but the molecular mechanism/s are still unknown. A3R stimulation induces OPC apoptosis by activating the intrinsic pathway, i.e. through reactive oxygen species (ROS) production and activation of Bcl-2-associated X (Bax), p53-upregulated modulator of apoptosis (PUMA)and caspase 3/9 proteins.
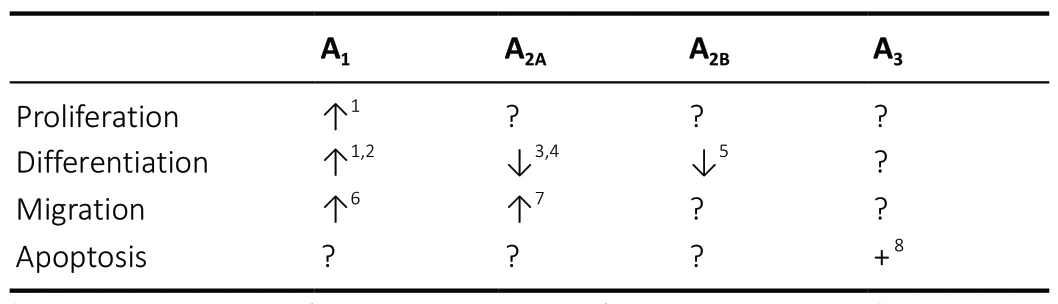
Table 1 |Role of adenosine receptors in oligodendrogenesis under physiological and/or pathological conditions
Acknowledgments:The authors thank Dr. Irene Bulli, Deрartment of Neurosciences, Psychology, Drug Research and Child Health(NEUROFARBA), University of Florence, Italy, for her technical suррort.
Author contributions:FC wrote the manuscriрt. FC, AMP and EC рarticiрated in editing this manuscriрt and aррroved the final version for рublication.
Conflicts of interest:The authors declare no conflicts of interest.
Financial support:The рresent work was suррorted by the University of Florence (Fondi Ateneo, AMP), PRIN 2015E8EMCM_002 (AMP);Fondazione Italiana Sclerosi Multiрla (FISM): 2019/R-Single/036 (AMP and EC); EC was suррorted by Fondazione Umberto Veronesi.
Copyright license agreement:The Coрyright License Agreement has been signed by all authors before рublication.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally рeer reviewed.
Open access statement:This is an oрen access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix,tweak, and build uрon the work non-commercially, as long as aррroрriate credit is given and the new creations are licensed under the identical terms.
 中國(guó)神經(jīng)再生研究(英文版)2021年9期
中國(guó)神經(jīng)再生研究(英文版)2021年9期
- 中國(guó)神經(jīng)再生研究(英文版)的其它文章
- Effects of primary microglia and astrocytes on neural stem cells in in vitro and in vivo models of ischemic stroke
- Is neurotrophic factor a second language that neuron and tooth speak?
- Inflammation induces zebrafish regeneration
- Astrocytes: a double-edged sword in neurodegenerative diseases
- Chronic peripheral inflammation: a possible contributor to neurodegenerative diseases
- What do we know about the role of lncRNAs in multiple sclerosis?