
Celiac disease:From genetics to epigenetics
2022-02-17 10:19:18ElisaGnodiRaffaellaMeneveriDonatellaBarisani
Elisa Gnodi,Raffaella Meneveri,Donatella Barisani
Abstract Celiac disease (CeD) is a multifactorial autoimmune disorder spread worldwide.The exposure to gluten,a protein found in cereals like wheat,barley and rye,is the main environmental factor involved in its pathogenesis.Even if the genetic predisposition represented by HLA-DQ2 or HLA-DQ8 haplotypes is widely recognised as mandatory for CeD development,it is not enough to explain the total predisposition for the disease.Furthermore,the onset of CeD comprehend a wide spectrum of symptoms,that often leads to a delay in CeD diagnosis.To overcome this deficiency and help detecting people with increased risk for CeD,also clarifying CeD traits linked to disease familiarity,different studies have tried to make light on other predisposing elements.These were in many cases genetic variants shared with other autoimmune diseases.Since inherited traits can be regulated by epigenetic modifications,also induced by environmental factors,the most recent studies focused on the potential involvement of epigenetics in CeD.Epigenetic factors can in fact modulate gene expression with many mechanisms,generating more or less stable changes in gene expression without affecting the DNA sequence.Here we analyze the different epigenetic modifications in CeD,in particular DNA methylation,histone modifications,non-coding RNAs and RNA methylation.Special attention is dedicated to the additional predispositions to CeD,the involvement of epigenetics in developing CeD complications,the pathogenic pathways modulated by epigenetic factors such as microRNAs and the potential use of epigenetic profiling as biomarker to discriminate different classes of patients.
Key Words:Celiac disease;Epigenetics;DNA methylation;Histone modifications;Long non-coding RNAs;MicroRNAs
lNTRODUCTlON
Celiac disease (CeD) is a multifactorial autoimmune enteropathy that develops in genetically predisposed subjects carrying the HLA-DQ2 or HLA-DQ8 haplotype.The prevalence of these HLA haplotypes in the general population is around 30%-40%,suggesting that they are necessary,but not sufficient,to induce CeD[1].Regional variancies have been observed in the frequency of these haplotypes in CeD,with the HLA-DQ8 that ranges from about 2%-4% in western countries to 25%-30% in the Iranian population and in the middle East[2].Indeed,CeD prevalence worldwide is about 1%-2%,with environmental factors that contribute to the regional differences,like the exposure to gluten,the known CeD exogenous antigen[1].The clinical presentation of CeD is quite heterogeneous,ranging from classical intestinal-related symptoms (diarrhoea,failure to thrive) to non-intestinal manifestations (anaemia,dermatitis,osteoporosis),and such diverse clinical picture may delay,in some cases,the prompt diagnosis[3].Even if the main mechanisms by which gluten peptides cause CeD intestinal lesion are now quite established,the reason why only a few of the genetically predisposed subjects develop the disease still needs to be clarified.In fact,geneticists calculated that the presence of a specific HLA accounted only for about 40% of the genetic predisposition,leaving most of the genes involved in the development of the disorder still unknown[4].Thus the initial focus of researchers was to identify further genes that could constitute a“genetic background”predisposing to the disease.Few studies used the classical linkage analysis approach,but the obtained results were specific of a single population and could not be replicated[5],or the identified genomic region did not harbour genes that appeared to be involved in CeD pathogenesis[6].The identification of additional predisposing genes was obtained using a different approach,i.e.,Genome Wide Association Studies (GWAS) performed on populations of different geographical origin.These studies identified 39 Loci associated with CeD development,but also confirmed the role of the Human Leukocyte Antigen (HLA) region.Additional analyses revealed that some loci included more than one gene associated with CeD,thus raising the number of the involved polymorphisms (single nucleotide polymorphisms,SNPs) up to 57.Although these studies were quite extensive,the identified loci were not able to completely explain CeD genetic predisposition,since HLA+all these additional genes accounted for about 54% of the hereditability of CeD[7].Interestingly,a great number of these SNPs involved genes with an immune function either in the intestine or in the thymus,further supporting the idea that an alteration of the immune response represents an essential step in CeD predisposition and pathogenesis.Moreover,a more recent paper was able,integrating genomic and transcriptomic data,to prioritize genes involved in CeD,and to identify TRAF-type zinc finger domain containing 1 (TRAFD1) as a master regulator of genes involved in interferon (IFN)γ signaling and MHC I antigen processing/presentation[8].The different combination of these SNPs could also be associated with a different phenotype,as recently reported by Cerquieiraet al[9],who identified the TLR7/ TLR8 Locus associated with disease onset before 7 years of age,whereas SH2B3/ATXN2,ITGA4/UBE2E3 and IL2/IL21 Loci were associated with later development of CeD and a more severe small bowel mucosal damage.In addition,SH2B3/ATXN2 was associated with type 1 diabetes;in fact some of the identified SNPs are described as predisposing factors also for other autoimmune disorders like type 1 diabetes and Crohn’s disease[7,10],almost suggesting the presence of a common genetic background predisposing to autoimmunity.It must also be noted that,among the identified SNPs,only 5% were present in coding regions,and about 5% and 9% in 5’ and 3’ untranslated regions,respectively[7].This means that 81% of the identified SNPs were either in intergenic or intronic regions,suggesting that their role could be to regulate gene expression,possibly through the interaction with transcription factors or proteins able to regulate chromatin status,i.e.,epigenetic modifications.Epigenetics has gained much attention in recent years,but its involvement in CeD still needs to be well characterized.First of all,this term has been used to define a wide range of concepts,and the 2008 Cold Spring Harbor meeting defined epigenetics as“the study of a stably heritable phenotype resulting from changes in a chromosome without alterations in the DNA sequence”[11].DNA methylations,histone modifications resulting in chromatin remodelling and non-coding RNAs are the three main categories that fall into this statement.The latter one also includes microRNAs,that have been found deregulated in CeD patients,suggesting that the regulation of the expression of target mRNAs by post-transcriptional modifications is relevant in CeD pathogenesis[12-14].In this direction,RNA sequencing studies are currently trying to identify different gene expression signatures that could help stratify patients based on CeD stage/presentation,or highlight new pathways implicated in CeD development[15-17].
In this review we will provide an update on the current knowledge about epigenetics in CeD and investigate the possible role of epigenetic profiling in patient classification and/or in determining the risk in subjects with familiarity to CeD.
LlTERATURE SEARCH
The literature search was carried out on PubMed,searching the key words“epigenetics”,“histones -acetylation,-methylation,-phosphorylation,-ubiquitination”,“chromatine”,“non-coding RNA”,“l(fā)ncRNA”,“miRNA”,“microRNA”,“RNA methylation”AND“celiac disease”.The reviewed papers were original research articles published from 2010 to October 2021.All articles that were considered relevant to the purpose of the present review were included,whereas the ones that did not add novelty or did not give clear results were excluded during the critical revision of the literature.
DNA METHYLATlON
DNA methylation is the most known epigenetic modification,that sees the family of DNA methyltransferase (DNMTs) enzymes transfer methyl-groups to the C-5 residue of cytosine pyrimidine ring.This phenomenon occurs in particular in CpG-rich regions (CpG islands),mainly localized in promoters and regulatory regions.Being the methylated DNA physically less accessible for transcription,this causes the inactivation of the nearby genes[18].The majority of gene promoters are normally methylated to guarantee a fine regulation of transcription in different tissues or cell types.Like most of the epigenetic modifications,it does not directly alter the DNA sequence,but a typical methylation profile can usually be inherited.Hypo-methylation or hyper-methylation of specific genomic loci has been reported to predispose to disease and cancer[19];for this reason,the first studies on DNA methylation in CeD analyzed the predisposition to develop small bowel adenocarcinomas.In particular,Bergmannet al[20] found a high level of CpGs methylation and microsatellite instability correlated to the loss of MLH1 expression in three different small bowel carcinomas in CeD patients,whereas this feature was not present in non-CeD patients.A similar finding was observed by Diosdadoet al[21],who also detected an hypermethylation of theAPCgene promoter that caused defects in the mismatch repair mechanisms in these patients.Further studies were conducted by Rizzoet al[22],who were able to identify four different CpG Island Methylator Phenotypes,with two of them being specific for CeD patients.
Although these data highlight a typical methylation pattern in CeD complications,the identification of a methylation profile associated with the predisposition for developing CeD is essential.Since GWAS pointed out the presence of many risk variants common to other autoimmune diseases,it has been hypothesised that they could also share common methylation patterns.Hammakeret al[23] focused on the specific risk variant rs906868,shared between CeD and rheumatoid arthritis and mapping in the promoter of LBH,gene with a regulatory function on the Wnt pathway,essential for differentiation and development.Interestingly,the authors detected a differential methylation pattern in the two diseases,thus suggesting the possibility of a disease-specific profile[23].Methylation can also occur only on one allele,thus not causing a complete silencing of the gene but its modulation.Hutchinsonet al[24] investigated allele-specific methylation (ASM) on CpG islands localized throughout the genome,detecting four of them as implicated in complex diseases.In particular,rs2762051,a C/T risk variant that maps in the non-coding RNA DLEU1,undergoes ASM in CeD[24].It is interesting to note that this variant belongs to the group of SNPs previously identified as significantly associated to CeD predisposition[7],thus further supporting the need for an integration between the genetic and epigenetic profile.A specific gene methylation could also be associated to a different phenotype,in particular if the methylation site is able to influence the expression of disease-related proteins.In fact,a lower level of methylation was detected in different IFNγ CpGs in patients with ulcerative colitis (UC) and a more severe phenotype[25],suggesting that methylation can regulate this UC pivotal pathogenetic pathway.Fernandez-Jimenezet al[26] demonstrated that this can also happen in CeD,since they observed not only a different methylation profile in the genes of the core NFkB pathway in active CeD subjects as compared to controls,but also an inter-mediate pattern in CeD patients on gluten-free diet (GFD).Although it was not clear if gliadin could directly affect the methylation of these specific genes,long exposure to gliadin could be able to affect the methylation pattern,since prolinerich peptides seem to act as opioid-like molecules causing a modulation in glutathione activity and DNA methylation[27].
It is also known that methylation profiles can be cell type-specific,thus a single cell methylation analysis could provide essential information.However,the methods employed to obtain single cell preparations can sometimes alter the DNA methylation patterns as described by Jenkeet al[28],thus pointing out the importance of considering this aspect in designing this kind of studies.Another potential bias of methylation studies regards the preferential amplification of an allele or a strand,based on its methylation degree.For this reason Ochoaet al[29] realized a new Bayesan calibration method and validated it also on CeD patients samples,and the most recent studies on methylation in CeD considered these techniques.Fernandez-Jimenezet al[30] compared the whole methylome of the epithelium and immune cells from CeD and controls biopsies,finding a cell-specific methylation pattern,with 43 (11 hypo-,32 hyper-methylated) and 310 (40 hypo-,270 hyper-methylated) differentially methylated positions (DMP) in the epithelium and in immune cells respectively.It is important to note that only a portion of the DMPs was shared between the two cellular components,highlighting a cell-specific,disease-driven modification.Cieloet al[31] used a different approach,separating the epithelium from the lamina propria and analysing the expression and methylation of genes known to be altered in CeD.Among the candidate genes,they found a decreased methylation in SB2H3 and IL21,that coupled with their increased expression in the epithelium and lamina propria respectively,but only in active CeD compared to controls.Since these genes take part in the pathways mainly involved in inflammation and barrier integrity in CeD,identifying a different signature in patients and controls may partially explain the abnormal response to food antigens and help patient stratification[31].Finally,the most recent study in this field by Hearnet al[32] compared methylation profiles in saliva samples obtained from CeD patients on a GFD and controls with the use of a beadchip array,finding a different methylation pattern in HLA-DQB1 and in specific loci nearSLC17A3gene in a pilot cohort,data not confirmed in a larger study group.Being this collection method noninvasive,it could be a great tool for patient discrimination,especially in the screening of predisposed subjects,once a different and discriminatory methylation profile has been clearly established.Moreover,the patient histological classification needs to be taken into account,even if a partial concordance in methylation patterns in saliva and intestine has been observed in a small group of patients[33].In this direction,encouraging results come from studies on inflammatory bowel diseases (IBDs) from Howellet al[34],that were able to statistically discriminate different classes of patients by analysing the transcriptome and the methylation status in the intestinal epithelium,correlating them with the disease outcome.Indeed,methylation profiling can be an asset to study different aspects of CeD,both to understand its pathogenesis and as a biomarker.A graphical representation of DNA methylation is present in Figure 1,whereas its highlights in CeD are reported in Table 1.
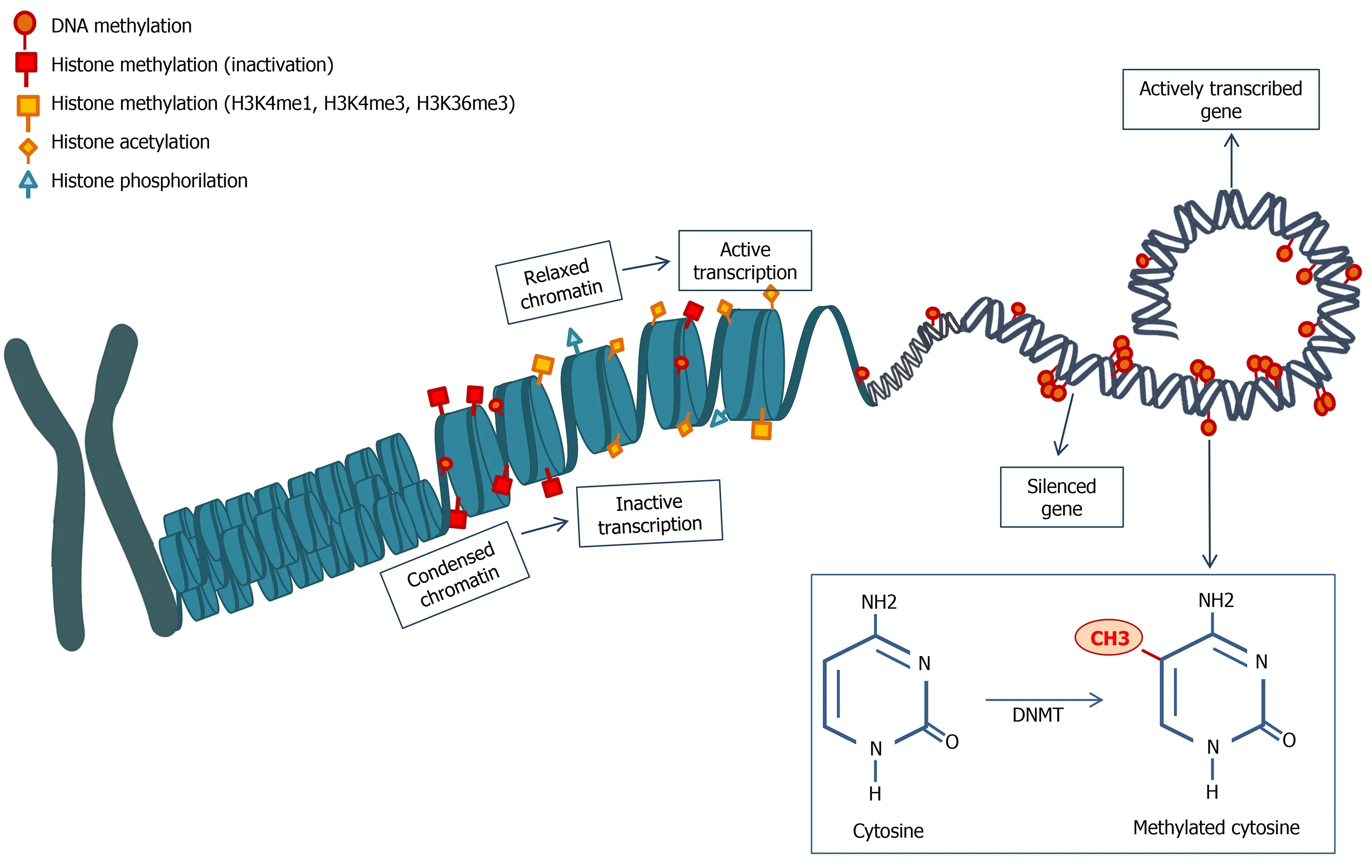
Figure 1 Schematic representation of DNA methylation and histone modifications.Histone modifications are many and determine a different chromatin status,modifying DNA accessibility and interfering with gene transcription.Methylation can also happen directly on the DNA sequence,mainly resulting in gene silencing.The image is original and was created with the use of Servier Medical Art modified templates,licensed under a Creative Common Attribution 3.0 Unported License (https://smart.servier.com).DNMT:DNA methyltransferase.
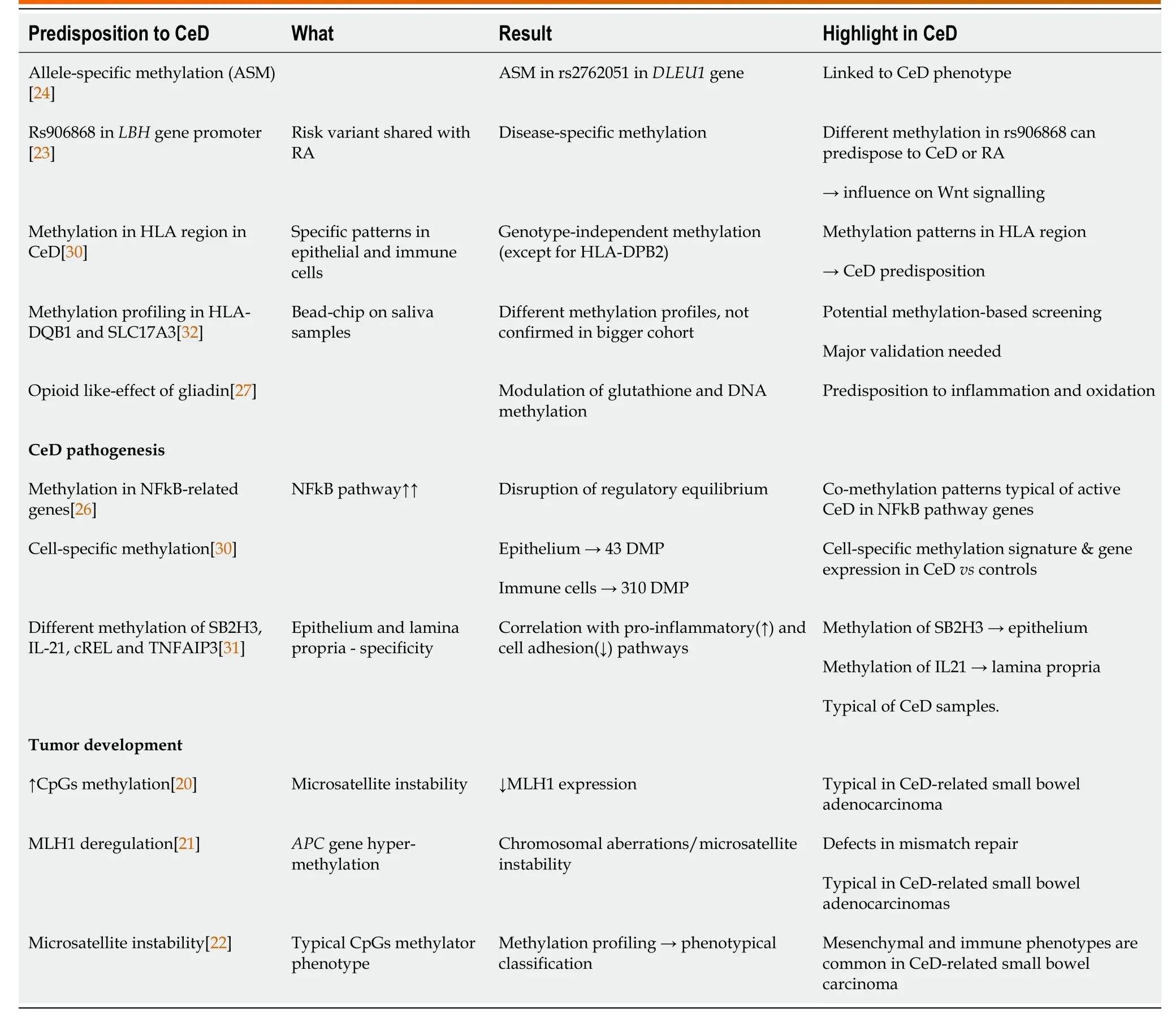
Table 1 DNA methylation features in celiac disease
HlSTONE MODlFlCATlONS
A further step in gene expression regulation in the nucleus is represented by histone modifications.Histones are key proteins of chromatin,able to modify its structure making the DNA more or less accessible to transcription.The most common histone modifications are acetylation,phosphorylation,ubiquitination and methylation.Gene expression analysis studies performed on CeD biopsies compared to controls,pointed out a differential regulation of histone-modifying enzymes,thus suggesting the presence of a disease-related epigenetic signature involving histone modifications[16,17].
Histone acetylation,governed by acetyltransferase (HATs) and deacetylase (HDACs) enzymes,usually result in active gene transcription due to chromatin relaxation[35].Zorroet al[36] analyzed the transcriptomic and epigenetic responses to IL-15,IFNβ and IL-21 stimulation in primary cytotoxic T lymphocytes derived from CeD biopsies.Specific transcriptomic patterns were identified for the different cytokine stimulation,but also different levels of histone acetylation (H3K27ac) were detected.Interestingly,the increase in transcription after IFNβ stimulation was associated,in about 60% of the cases,with an increase in H3K27ac in promoter and enhancer regions,whereas this was not the case after IL-15 stimulation,pointing towards the presence of a different regulatory mechanism.In fact,upon IL-15 stimulation a relevant number of differentially expressed genes were ncRNAs,suggesting their potential regulatory role independently from H3K27ac modifications[36].
Histone methylation is more complex and mainly linked to repression,but the specific methylations H3K4me1,H3K4me3 and H3K36me3 are commonly found in actively transcribed regions[35].In fact,H3K4me3 was used as a marker of active transcription by Gutierrez-Achuryet al[37];the authors firstly identified SNPs associated with both CeD and rheumatoid arthritis,but also SNPs specific for each disease.The combination of these data with H3K4me3 profiles available in public databases allowed the authors to detect similar histone enrichment corresponding to the common SNPs in less specialized immune cell types,whereas disease-specific SNPs overlapped with H3K4me3 profiles in more specialized cells[37].H3K36me3 signature was investigated by Moffittet al[38] in the enteropathy associated T cell lymphoma (EATL),the most common neoplastic consequence of CeD.The histonelysine N-methyltransferase SETD2 is the responsible for this kind of methylation and it was silenced in 32% of EATL in CeD subjects.In fact,in vivostudies with a knock out model for SETD2 in T cells carried a decreased H3K36me3 pattern along with γδ-T cells expansion[38].H3K27me3 was instead used as gene silencing signature by Oittinenet al[39] in determining the involvement of the polycomb repressive complex 2 (PRC2) in controlling Wnt signalling in the intestine.They found a H3K27me3 pattern which differed according to the cell position/differentiation along the crypt/villus axis,and involved also genes related to proliferation and differentiation in the epithelium.Thus PRC2-driven tri-methylation is important to maintain the homeostasis driven by Wnt;however,since Wnt pathway deregulation has been connected to CeD pathogenesis,they also hypothesise that a Wnt/PRC2 disrupted axis could cause the development of crypts hyperplasia in CeD[39] (Table 2).However,among the external factors that can influence the epigenetic regulation,the interaction with the microbiota needs also to be considered,since changes in the gut bacterial populations can affect the activity of enzymes involved in epigenetic regulation[40,41].
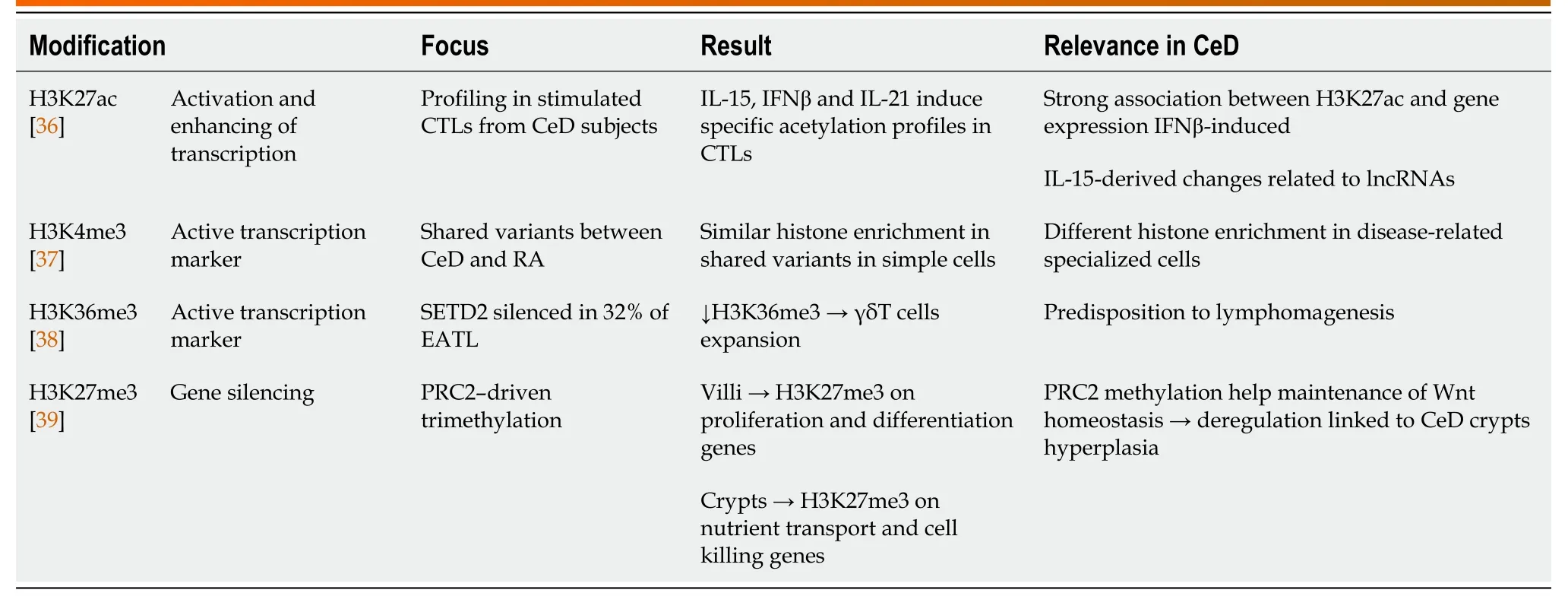
Table 2 Celiac disease-relevant histone modifications
LONG NON-CODlNG RNAS
Long non-coding RNAs (lncRNAs) have emerged in recent years as a class of transcripts with a wide spectrum of mechanisms of action that can affect gene expression regulation both at transcriptional and post-transcriptional level,in the nucleus and in the cytosol.Their genomic location is various,either being intergenic or within the introns of coding genes.Among their many mechanisms of action,the most frequent are direct DNA sequence interaction,transcription factor sequestration,chromatin rearrangement,regulation of histone modifications and microRNA sponging (Figure 2)[42,43].Being implicated in so many regulatory patterns,they have been studied in cancer and in many other diseases,also with an autoimmune background[44].
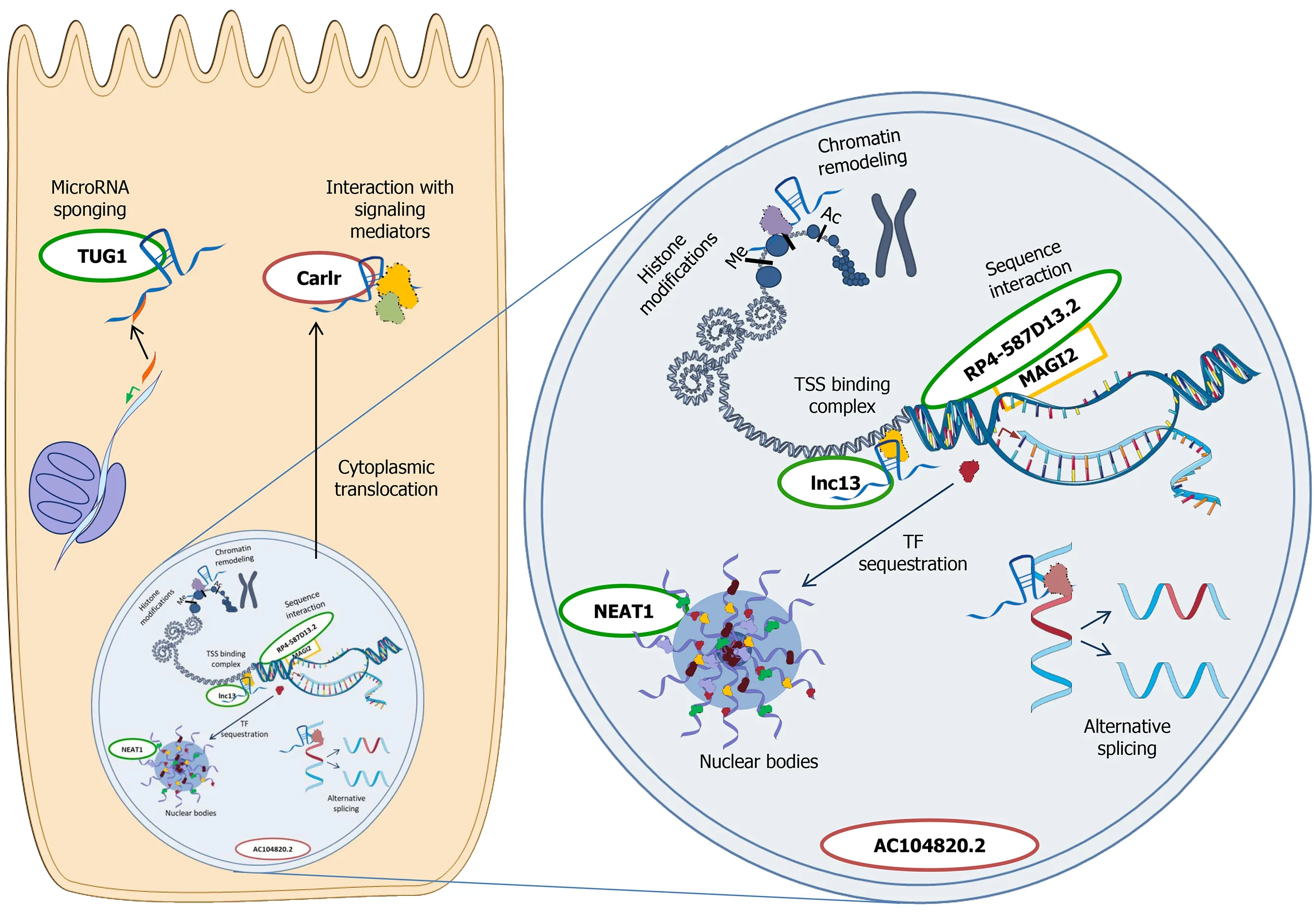
Figure 2 Main mechanisms of action of long non-coding RNAs in the cell.Cytoplasm -microRNA sponging:microRNAs are sequestered from their mRNA targets,resulting in mRNA translation;Interaction with signalling mediators:binding to pathway mediators can modulate downstream targets activation.Nucleus -chromatin remodeling:Long non-coding RNAs (lncRNAs) interact with chromatin and modify its conformation;histone modifications:lncRNAs influence the activity of the enzymes responsible for histone modifications;sequence interaction:lncRNAs act on nearby genes transcription;transcription start site (TSS) binding complex:lncRNAs bind ribonucleoproteins,interfering with gene transcription;transcription factor (TF) sequestration in nuclear bodies:TF are moved to nuclear bodies from the promoter region of target genes,influencing gene transcription.Nuclear bodies like paraspeckles consist in a lncRNA scaffold and target proteins;alternative splicing:lncRNAs determine a preferential splicing in favour of an isoform respect to another one.The so-far investigated lncRNAs in celiac disease (CeD) are reported near their known mechanism of action,respecting their main cellular localization.lncRNAs in green were reported to be downregulated in CeD,whereas the ones in red were found upregulated in a specific cell compartment.The image is original and was created with the use of Servier Medical Art modified templates,licensed under a Creative Common Attribution 3.0 Unported License (https://smart.servier.com).TSS:Transcription start site;TF:Transcription factor.
The pathogenesis of CeD involves several different mechanisms,i.e.,the passage of the gluten peptides across the intestinal barrier,but also the activation of innate and adaptive immune response.LncRNAs can influence all these processes,but the expression of these RNAs can,in turn,be affected by the presence of SNPs.
A typical aspect in CeD pathogenesis is the loosening of the tight junctions (TJ),that increases the intestinal permeability[45].Jauregi-Miguelet al[46] identified,in a genomic region associated with CeD,a lncRNA named RP4-587D13.2.This is located in an intron ofMAGI2gene,a scaffold protein present in the tight junction plaque and able to regulate epithelial integrity.Interestingly,RP4-587D13.2 can regulate MAGI2 expression,altering the downstream TJ-related proteins like CLDN1 and ZAK.RP4-587D13.2 and MAGI2 resulted also downregulated in CeD samples (both with active CeD and on a GFD) and it seems that gliadin stimulation can reinforce this effects[46].
Rica?o-Ponceet al[47] identified genes located in close proximity to autoimmunerelated SNPs,revealing that 42 of these SNPs could specifically affect the expression of 53 non-coding RNAs.In particular,two lncRNAs were associated to CeD-specific SNPs,namely AP002954.4 and AC104820.2.In both cases the SNPs were able to affect the expression of these lncRNAs,which had a role in the immune response[47].Interestingly,AC104820.2 had already been detected upregulated in active CeD patients intestinal mucosa by Plaza-Izurietaet al[48],who also identified the SNP rs1018326 as associated with this lncRNA.These data thus support the involvement of lncRNAs in the immune response in CeD,however there are aspects that must be kept in mind,as also reported by Hrdlickovaet al[49].In fact,although the authors suggested the lncRNAs RP3-395 M20.9 and IL21-AS to be relevant in CeD,and linked them to TNF and IL-21 pathways,they also reported that the same lncRNAs are involved in different autoimmune disorders.In addition,some lncRNAs were detected only in some specific cell subtypes,underlying the need for a cell-specific analysis[49].
Castellanos-Rubioet al[50] identified lnc13 as associated to the susceptibility to celiac disease,demonstrating also the functional role of this lncRNA.Lnc13 binds the nuclear ribonucleoprotein hnRNPD and histone deacetylase 1,forming a complex that acts as a repressor of inflammatory gene expression.The presence of inflammatory stimuli activate the NFkB pathway that reduces lnc13,thus removing the repression on gene transcription.Interestingly,in CeD patients,not only lnc13 expression is reduced,but there is also a SNP that generates a lnc13 variant that binds hnRNPD less effectively,possibly contributing to CeD development[50].NFkB activation is central in CeD-driven inflammation,and it is able to induce the expression of another lncRNA,i.e.,Carlr.This molecule is localized in the nucleus,but it translocates in the cytosol after the activation of the NFkB pathway,and it seems essential for the induction of downstream genes such as IL-1β in macrophages.However,differently from what expected,Carlr was downregulated in total biopsies from active CeD patients,although its cytosolic fraction was increased;this observation thus needs further evaluation to determine the real role of this lncRNA in the intestinal tissue[51].
Finally,it is known that IL-15 production has a central role in CeD pathogenesis[52].Zorroet al[36] in their work highlighted a subset of ncRNAs that were downregulated by IL-15 stimulation,suggesting the involvement of this class of transcripts in IL-15 response.A recent study by our group demonstrated that gliadin induces TUG1 and NEAT1 expression in biopsies obtained from CeD patients on gluten-free diet,mechanism that depends on innate immunity activation in the case of NEAT1.In fact,IL-15 can trigger NEAT1 expression bothex vivoand in vitro,through the newly identified axis IL-15/STAT3/NEAT1[53].The effects of NEAT1 upregulation deserve more investigation,since this lncRNA localizes in the nucleus and is able to bind transcription factors,thus sequestering them and,in turn,regulating the expression of several genes,such as IL-8[54].
RNA METHYLATlON
A further regulation can be played by RNA modifications.In fact,recent findings are pointing out that direct sequence methylation can also happen at RNA level.Differently from DNA,RNA can undergo many more kinds of methylation (up to 72),involving both mRNA and non-coding RNAs.Previous studies found RNA methylation linked to biological and immune processes,as well as complex pathologies like obesity[55].Up to now,the main limitation in RNA methylation studies was the difficulty to perform a site-specific detection and quantification.Using a recently developed method that allows to detect m6A,the most frequent RNA modification,Olazagoitia-Garmendiaet al[56] characterised the CeD-associated SNP rs3087898,located in the 5’ untranslated region of exportin1 gene,a modulator of NFkB pathway.The mRNA derived from the CeD-associated variant was preferentially methylated with respect to the normal one,and this modification was able to increase the protein production,resulting in an inflammatory microenvironment.Besides,gliadin was able to increase this modification and exportin1 protein levels,bothin vitroand in CeD patients samples[56].Altogether,these studies suggest that attention should be given also to this kind of modifications when focusing on gene expression regulation in CeD.
MlCRORNAS
MicroRNAs (miRNAs) are short RNA sequences (20-25 nucleotides) accounting for about 30% of gene expression regulation,affecting various processes,including chromatin conformation and transcription,as well as mRNA stability and translation.Usually they present an inverse relationship with their targets,inducing their downregulation.
miRNAs can also be detected in the plasma,and for this reason they have been evaluated in a large number of different disorders,in the attempt to identify specific biomarkers for early diagnosis or follow up.Initial papers on CeD screened the duodenal tissue to identify a specific signature representative of the events in the mucosa,and to determine whether the“signature”miRNAs were returning to normal levels in patients on GFD.Capuanoet al[57] evaluated biopsies of children with CeD at diagnosis or on GFD,identifying about 20% of the analyzed miRNAs as differentially regulated.The authors then focused on the upregulation of miR-449a[57],which targets Notch1 and KLF4,genes involved in intestinal cell proliferation and differentiation.Indeed Notch1 expression was significantly downregulated in CeD biopsies,confirming the regulatory role of miRNAs also on proliferation in CeD.Other miRNAs were also downregulated,i.e.,miR-124a,miR-189,miR-299-5p and miR-379,similarly to what reported in Crohn’s disease[58].
Studies on adult CeD patients identified different subset of miRNAs according to the clinical picture or the severity of intestinal damage[12,14].A significant downregulation of miR-31-5p was observed in all the CeD groups,whereas miR-192-3p and miR-192-5p were downregulated in CeD patients with anemia and a severe histological lesion,respectively.miR-192-5p targeted two different molecules involved in the innate immunity,i.e.,NOD2 and CXCL2,that were upregulated in CeD patients,in particular in severe cases[12].On the other hand,miR-31-5p had as a target Foxp3,essential for Treg development;again,a significant inverse correlation was observed between the miRNA and the target mRNA.These changes were induced by gliadin,as demonstrated byin vitrostimulation of biopsies of patients on GFD[12].It is interesting to note that the data on miR-192 are similar to those observed in IBDs,in which miR-192 regulated NOD2 expression[59].
However,a more comprehensive approach was needed,and a recent paper correlated,in the duodenal mucosa of celiac subjects,the miRNA and mRNA expression patterns obtained by sequencing[17].The data analysis revealed the presence of a complex network involving various pathways known to be deregulated in CeD,such as immunity (interferon),suggesting that CeD-associated miRNAs play a central role in causing the intestinal damage.
The identification of a specific miRNA plasma signature remains elusive at the present time.An analysis performed in pediatric CeDs revealed the presence of a trend similar to that observed in duodenal biopsies for miR-192-5p,miR-31-5p and miR-21-5p.However none of these miRNAs could be defined as a marker able to evaluate the recovery of the mucosa on GFD,since there was no return to normal levels (miR192-5p) or a clear cut-off could not be established (miR-31-5p and miR-21-5p)[13].A further study performed by Amret al[60] analyzed the same miRNAs,i.e.,miR-21 and miR-31,identifying a cutoff value and determining a sensitivity and specificity of 82.4%and 80.8% for miR-21 and 93.8% and 72% for miR-31.Another recent paper confirmed the value of plasmatic miR-21 as a marker,with a sensitivity and specificity of 0.65 and 0.83,but better results were obtained considering miR-155 (0.94 and 0.87,respectively)[61].Although encouraging,these results need further validation in larger cohorts;moreover even for miR-21 the identified cutoff values were quite dissimilar,thus suggesting the need to determine the cutoff according to the method and equipment employed for the analysis.Last but not least,the data obtained in CeD patients will have to be compared with those obtained from subjects with other inflammatory disorders such as IBDs,to confirm the specificity of the findings.
MACHlNE LEARNlNG
As described,the evaluation of the epigenome generates large datasets,and the data increases when the integration of different datasets (such as those derived from methylome and long/short non-coding RNA transcriptome) is required.The generation of a large quantity of data allows to move towards a data-driven analysis,i.e.,machine learning or artificial intelligence (these terms are now regarded as interchangeable).Machine learning can,through statistical methods and algorithms,make classifications or predictions of input data,such as recognize a specific endoscopy pattern and classify the patient as celiac or non-celiac.In simpler machine learning approaches supervised learning is commonly used,i.e.,the use of labeled datasets designed to train or“supervise”algorithms into classifying data or predicting outcomes.In the medical field this obviously requires training datasets selected by specialized clinicians (for example the identification of endoscopic images characteristic of celiac disease).Deep learning is a subset of machine learning,in which the algorithms are more complex and include more than one layer in the neuronal network.For deep learning the presence of a labeled training dataset is not required,since these algorithms can analyze data in the raw form and determine the features that distinguish the various groups.
With regards to CeD,the machine learning approach has been originally used to analyze endoscopic data,either those generated by upper endoscopy[62] or by videocapsule and,in this latter case,the deep learning approach was able to reach a high sensitivity and specificity in the diagnosis of CeD using the video capsule images[63,64].Although these data seem quite encouraging,it must be kept in mind that the gastrointestinal tract is a complex environment,and several factors can affect the obtained images,as discussed in depth by Hegenbartet al[65].The deep learning approach could be useful also in the evaluation of duodenal biopsies,as demonstrated by different authors that were able to develop artificial intelligence-based methods for the correct classification of duodenal samples[66-68].Machine learning has also been employed to improve the diagnosis based on B/T cell repertoire[69,70];although the data obtained in the latter case are quite interesting,this approach is not easily applicable for routine diagnosis.
A machine learning approach could have a very important clinical application should it be able to predict who,within a risk population,will develop the disease.An encouraging study that analyzes this aspect has recently been published by Piccialliet al[71],who tested different models in order to predict the development of an overt CeD in childen with potential CeD.The use of a machine learning approach towards basic science data obtained from duodenal biopsies of CeD patients has recently been published by Wolfet al[16],who analyzed not only the trascriptomic profiles but also the expression of some lncRNAs and miRNAs.Moreover,the authors focused on gene expression signatures which associate with transcription factor (TF)-activity and chromatin state,as well as DNA and histone methylation pattern,thus providing an indirect measure of epigenetic modifications.This comprehensive analysis revealed,for example,a different role of miRNAs and lncRNAs in the regulation of gene expression,correlating the latter category with more subtle adjustments of the transcription under control of epigenetic mechanisms.
CONCLUSlON
Understanding the epigenetics of celiac disease remains mandatory in order to completely clarify the pathogenetic processes behind its development and the spectrum of its manifestations.The data here reviewed demonstrate that epigenetic changes occur in celiac disease,however further data are still needed before the identification of these changes could be used to screen or better categorize the patients,as depicted in Figure 3.In particular it will be essential to verify that the characteristics detected at a peripheral level (i.e.,blood or saliva) correspond to those present in the intestinal mucosa.In this regard,data seem encouraging for miRNAs,but the identification of a specific signature will probably require the combination of more than one of them to provide an excellent sensitivity and specificity.
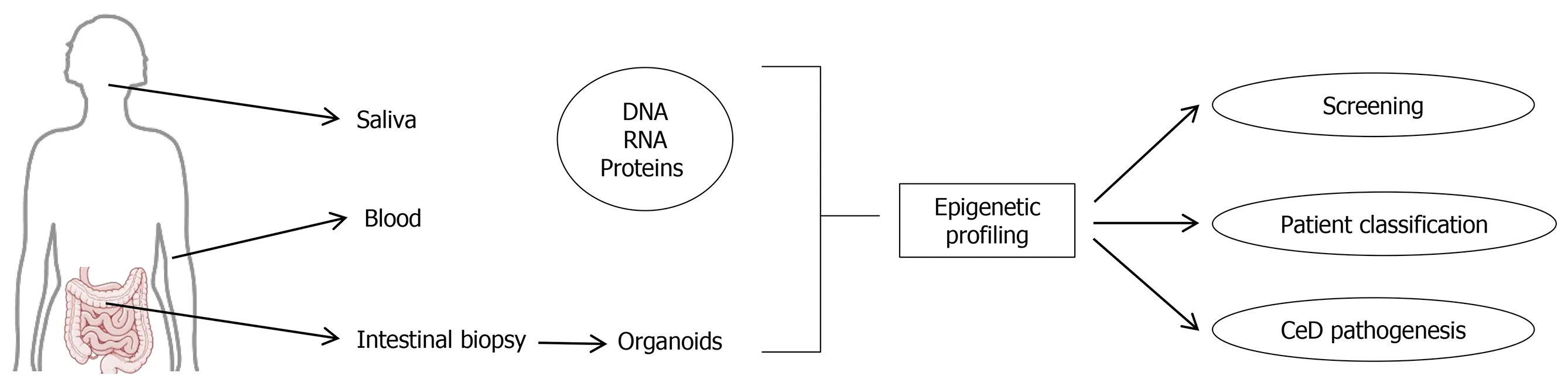
Figure 3 Potential use of epigenetics studies in celiac disease.The image is original and was created with the use of Servier Medical Art modified templates,licensed under a Creative Common Attribution 3.0 Unported License (https://smart.servier.com).CeD:Celiac disease.
ACKNOWLEDGEMENTS
We do thank Chris Kenyon,PhD for his suggestions regarding the machine learning approach.
 World Journal of Gastroenterology2022年4期
World Journal of Gastroenterology2022年4期
- World Journal of Gastroenterology的其它文章
- ls CA19-9 effective in predicting chemotherapeutic response in patients with synchronous liver metastases with colorectal cancer?
- lnterplay between chronic hepatitis B and atherosclerosis:lnnovative perspectives and theories
- Fibrinogen-like protein 2 deficiency inhibits virus-induced fulminant hepatitis through abrogating inflammatory macrophage activation
- Knockdown of DEAD-box 51 inhibits tumor growth of esophageal squamous cell carcinoma via the Pl3K/AKT pathway
- Sarcopenia in hepatocellular carcinoma:Current knowledge and future directions
- Gut bless you:The microbiota-gut-brain axis in irritable bowel syndrome