
Protein synthesis modulation as a therapeutic approach for amyotrophic lateral sclerosis and frontotemporal dementia
2022-11-05 14:29SantiagoCharifFlorenciaVassalluLaraSalvaalLionelIgaz
中國神經(jīng)再生研究(英文版) 2022年7期
Santiago E. Charif, M. Florencia Vassallu, Lara Salva?al, Lionel M. Igaz
Abstract Protein synthesis is essential for cells to perform life metabolic processes. Pathological alterations of protein content can lead to particular diseases. Cells have an intrinsic array of mechanisms and pathways that are activated when protein misfolding, accumulation,aggregation or mislocalization occur. Some of them (like the unfolded protein response)represent complex interactions between endoplasmic reticulum sensors and elongation factors that tend to increase expression of chaperone proteins and/or repress translation in order to restore protein homeostasis (also known as proteostasis). This is even more important in neurons, as they are very susceptible to harmful effects associated with protein overload and proteostatic mechanisms are less effective with age. Several neurodegenerative pathologies such as Alzheimer’s, Parkinson’s, and Huntington’s diseases, amyotrophic lateral sclerosis and frontotemporal dementia exhibit a particular molecular signature of distinct, unbalanced protein overload. In amyotrophic lateral sclerosis and frontotemporal dementia, the majority of cases present intracellular inclusions of ubiquitinated transactive response DNA-binding protein of 43 kDa (TDP-43). TDP-43 is an RNA binding protein that participates in RNA metabolism, among other functions. Dysregulation of TDP-43 (e.g. aggregation and mislocalization) can dramatically affect neurons, and this has been linked to disease development. Expression of amyotrophic lateral sclerosis/frontotemporal dementia TDP-43-related mutations in cellular and animal models has been shown to recapitulate key features of the amyotrophic lateral sclerosis/frontotemporal dementia disease spectrum. These variants can be causative of degeneration onset and progression. Most neurodegenerative diseases(including amyotrophic lateral sclerosis and frontotemporal dementia) have no cure at the moment; however, modulating translation has recently emerged as an attractive approach that can be performed at several steps (i.e. regulating activation of initiation and elongation factors, inhibiting unfolded protein response activation or inducing chaperone expression and activity). This review focuses on the features of protein imbalance in neurodegenerative disorders and the relevance of developing therapeutical compounds aiming at restoring proteostasis. We strive to highlight the importance of research on drugs that, not only restore protein imbalance without compromising translational activity of cells, but are also as safe as possible for the patients.
Key Words: amyotrophic lateral sclerosis; frontotemporal dementia; neurodegeneration;neurodegenerative diseases; protein imbalance; protein synthesis modulation;proteostasis; therapeutical compounds; transactive response DNA-binding protein of 43 kDa; translation; unfolded protein response
Introduction
Protein synthesis homeostasis allows cells to carry out normal processes that are necessary for life as we know it. Since proteins can have diverse functions (structural, signaling,receptor, connective, defensive, catalytic, etc.) and they need to be continuously produced and usually readily available, the molecular machinery of the cell must tightly control key steps of the synthesis process. These stages include verification of proper protein folding (as three-dimensional conformation is directly responsible for its function), re-folding or elimination if this is not achieved (Ciechanover and Kwon, 2017). As these steps are crucial to avoid the production of abnormal proteins,there must be a system in place to alert when an overload of misfolded or aberrant proteins is present.
The formation of misfolded protein aggregates is a hallmark of several neurodegenerative proteinopathies, causing toxicity due to altered protein homeostasis (proteostasis),among other reasons. Proper protein synthesis is monitored through cellular mechanisms involved in refolding and stabilizing these polypeptides (i.e., chaperone proteins),degradation (i.e., the ubiquitin-proteasome system [UPS])or, if the insult is significant, activation of molecular pathways that halt translation until the problem is solved(i.e. unfolded protein response or UPR) (Martinez et al.,2018). This multistep control network ensures that only properly folded and functional proteins are present in healthy cells. The consequences of chronic proteostasis stress in a neurodegenerative disease context may include brain atrophy,neuronal loss, impaired motor, and cognitive functions, and in some cases, death or chronic disability (Suresh et al., 2018).Most neurodegenerative diseases like Parkinson’s disease (PD),amyotrophic lateral sclerosis (ALS), frontotemporal dementia(FTD), Huntington’s disease, and Alzheimer’s disease (AD)can be classified as protein misfolding disorders. All of them involve the aggregation of unfolded or misfolded pathological proteins and defects in the systems that eliminate them. The manifestation of all these diseases is quite different, but they all have in common the accumulation of an abnormal quantity of unfolded proteins (Elmatboly et al., 2020). In recent years,the modulation of neuronal translation has gained clinical interest. This is evidenced by the development of new,effective drugs that act on key regulators of proteostasis and translation (Halliday et al., 2015; Sidrauski et al., 2015a, b;Vieira et al., 2015; Halliday et al., 2017; Mercado et al., 2018;Wong et al., 2019). The vast majority of neurodegenerative disorders can progress very fast and remain without cure.Treatments are mostly palliative or they result in a short lifespan extension. This fact underscores the need to discover novel compounds that regulate some of the factors altered in disease, in order to mitigate the translational repression or protein overload effects.
Data Sources
Electronic literature search was performed using PubMed(NCB?, USA) database, repositories such as Semantic Scholar(Allen ?nstitute for Artificial ?ntelligence, USA), Europe PubMed Central (EMBL-EBI, UK), JSTOR (USA), and the Google Scholar search engine (USA). Various combinations of the following keywords were employed for searching and screening of relevant information: protein balance, UPR, proteostasis,neurodegenerative diseases, protein synthesis modulation,FTD, ALS, TDP-43, translation, drugs that modulate translation,therapeutical compounds. As a general example, PubMed search for “drugs that modulate translation” was carried out covering 10 years of literature, excluding meta-analyses and including clinical trials, peer-reviewed publications, and reports. Eligibility involved research on nervous system,both done on in vitro/in vivo models or in patients with neurodegenerative disease. The last date searched was March 1, 2021.
Maintenance of Proper Protein Balance in the Cell
The process of protein synthesis is highly regulated and the correct folding of proteins is critical for cellular homeostasis.The cell possesses an array of mechanisms to preserve the correct folding and location of proteins in order to perform their function and, in that way, maintain proteostasis (McAlary et al., 2020). When cellular protein levels are unbalanced,different molecular mechanisms responsible for restoring homeostasis are recruited. When they fail, proteostasis is severely compromised and specific cellular responses that involve inhibition of global or local protein synthesis might be triggered. Newly synthesized proteins are folded by chaperone proteins, which generally interact with the hydrophobic residues of the polypeptides in order to avoid their interaction with the water present in the cytoplasm (Dahiya and Buchner,2019). When proper or complete folding is not achieved,chaperones can activate different cellular programs to deal with the misfolded proteins and restore proteostasis. These programs comprise the UPS, the UPR, and the heat shock response, among others (Hohn et al., 2020). Autophagy is another key pathway involved in both the removal of misfolded/aggregated proteins and damaged organelles(Mputhia et al., 2019).
UPS activation via the endoplasmic reticulum (ER)-associated degradation involves a joint work between the ER and UPS to mark the proteins, take them to the cytoplasm and degrade them. Ubiquitination is a 3-step process in which a protein is targeted to the proteasome and degraded. Once the protein is in the proteasome, the ubiquitin molecules can be recycled.Conversely, during autophagy, the misfolded protein is eliminated from the cell in lysosomes, specialized vesicles that carry hydrolytic enzymes (Klaips et al., 2018).
The UPR consists of three pathways involving transmembrane proteins that sense misfolded proteins in the lumen of the ER.These are the inositol-requiring enzyme alpha (?RE1α), PRKlike ER kinase (PERK), and the activating transcription factor 6 (ATF6). When unfolded proteins reach a threshold, both PERK and ?RE1α are activated via oligomerization and transautophosphorylation, whereas ATF6 translocates to the Golgi complex (Benedetti et al., 2000). ?RE1α functions as a cytosolic endoribonuclease that specifically cleaves an intron from the transcription factor X-box binding protein 1 transcript. This event generates an open reading frame that is translated into a protein (X-box binding protein 1s) that acts as a transcription factor, leading to the expression of multiple genes related to elements of the UPR and the integrated stress response.When adaptation through the splicing of X-box binding protein 1 fails and protein imbalance persists, ?RE1α cuts some specific microRNAs that are in charge of repressing the translation of the pro-apoptotic protein caspase-2. Thus, the levels of this protein increase and the mitochondrial apoptotic pathway is triggered (Upton et al., 2012). On the other hand,PERK is a cytosolic kinase that regulates translation through phosphorylation of the eukaryotic translation initiation factor alpha (e?F2α). ?n this way, a reduction in the total amount of proteins produced in the cell also decreases the burden of folding proteins until homeostasis is restored. In order to reactivate translation, the guanidine nucleotide exchange factor e?F2B replaces the GDP in e?F2α. ?f e?F2α is phosphorylated, e?F2B is inhibited due to the stabilization of eIF2-GDP, leading to general inhibition of protein synthesis and to the generation of stress granules (SG) containing inactive translation initiation complexes (Rabouw et al., 2019).When stress becomes chronic and e?F2α dephosphorylation-involved in the restoration of protein synthesis- fails, PERK induces the synthesis of ATF4 (a transcription factor that stimulates cell recovery) and CHOP. Finally, these transcription factors generate a cascade that culminates in BAK/BAXdependent apoptosis (Costache et al., 2012).
Proteostasis is even more relevant in post-mitotic cells like neurons, where these processes become less effective with aging and result in the accumulation of misfolded proteins. This accumulation of proteins, along with a poor set of mechanisms required to deal with oxidative stress,results in a greater susceptibility to the development of neurodegenerative diseases (Hohn et al., 2020). Protein inclusions are a hallmark of many neurodegenerative diseases. Examples include the β-amyloid composed plaques and tangles of phosphorylated tau in AD and α-synuclein aggregates in PD. Also, ubiquitinated inclusions are present in motor neurons in ALS (Farrawell et al., 2020). TDP-43 has been identified as the main component of ubiquitinated inclusions in sporadic ALS and in both familiar and sporadic FTD (Neumann et al., 2006). Importantly, the formation of these aggregates of misfolded TDP-43 in the cytoplasm is accompanied by the depletion of TDP-43 from the nucleus.
Altered Protein Balance and Neurodegenerative Diseases
Neurodegenerative disease is a term used to describe a group of disorders characterized by motor and cognitive deficits due to the loss of neurons. Many of these diseases present protein aggregates that contribute to a higher level of oxidative stress within the cell which, in turn, may lead to apoptosis. In the case of PD, the presence of Lewy bodies in the dopaminergic neurons of patients’ brains is a pathological hallmark. Lewy bodies are large aggregates of α-synuclein, an intrinsically disordered protein of 140 amino acids that is expressed in the brain and is described to have synaptic functions. In control subjects, only a small portion is phosphorylated in Ser129 under normal physiological conditions, but in Lewy bodies of PD patients, this proportion reaches almost 90%.So, hyperphosphorylation is hypothesized to be related with the aggregation of α-synuclein (Amanullah et al., 2017).The brains of AD patients usually present extracellular β-amyloid plaques and intracellular neurofibrillary tangles composed of hyperphosphorylated tau protein. The latter contains 85 possible phosphorylation sites and, when hyperphosphorylated, tau loses its ability to bind correctly to the microtubules, eventually leading to neuronal dysfunction and neurodegeneration. AD can be also produced by the mutation of the gene for amyloid precursor protein or in presenilin genes, both critical for amyloid generation and metabolism (Kurtishi et al., 2019).
Altered Proteins in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia
ALS is a progressive and fatal disease characterized by a loss of motor neurons in the spinal cord and the brain leading to muscle weakness. Most cases are sporadic, but a smaller percentage of cases are familiar, being both clinically indistinguishable. FTD is the second most frequent cause of dementia with an early onset, showing an average age of presentation between 45 to 65 years old. In this disease,deposition of tau protein or TDP-43 inside of the neurons is mainly associated with loss of neurons, glia, and changes in frontal lobes, anterior temporal lobes, and anterior cingulate cortex. Currently, three clinical subtypes of FTD are described, with two variants affecting the language (semantic dementia and progressive nonfluent aphasia) and a behavioral variant (bvFTD) that shows apathy, compulsive behaviors,and disinhibition among other symptoms (Devenney et al.,2019). To this date, several genes have been identified as responsible for the development of familiar ALS, such as FUS, SOD, C9ORF72, and TARDBP. Physiologically localized in the nucleus of the neuron, the RNA/DNA binding protein FUS participates in multiple cellular processes. The mutant form is primarily localized in the cytoplasm of the neuron,forming inclusions typically in the dentate gyrus (Kwiatkowski et al., 2009; Younes and Miller, 2020) and represents 4% of familial ALS cases and less that 1% of sporadic cases. Under normal conditions, SOD1 localizes in the cytoplasm reducing superoxide levels, while mutations in this protein give rise to cytotoxic properties (Rosen et al., 1993). Around 20% of familial cases of ALS present SOD1 mutations, while near to 2% are sporadic cases (Goutman et al., 2018). ?n physiological conditions, TDP-43 is localized within the nucleus and is involved in transcript regulation, stabilization, and transport,among other processes. Under pathological conditions, TDP-43 is found in the cytoplasm forming inclusions of misfolded protein. The presence of aggregates composed of insoluble TDP-43 is the most frequent pathological hallmark of both sporadic and familial ALS.
In FTD, the most relevant genes related to the development of the pathology are microtubule-associated protein tau responsible for 20–30% of familial cases of FTD (Couratier et al., 2017), C9ORF72 (described below), and progranulin. These genes exhibit an autosomal dominant pattern of inheritance and the last two are associated with TDP-43 pathology.
Tau is a microtubule-associated protein that regulates cytoskeletal turnover, particularly in axons. Mutated versions,generated by missense mutations and deletions or by the altered ratio of isoforms, show enhanced propensity to aggregate, associated with filamentous inclusions. This affects axonal transportation and microtubule dynamics, leading to neurodegeneration (Young et al., 2018). Progranulin is a secreted protein involved in the regulation of inflammation,wound repair, and development. Mutation in this gene causes haploinsufficiency (Baker et al., 2006) and it is present in 10–20% of familiar cases. Haploinsufficiency results in reduced progranulin levels, enhancing neuroinflammation and further neurodegeneration (Baker et al., 2006). Remarkably,homozygous loss of its expression causes a lysosomal storage disorder termed neuronal ceroid lipofuscinosis (Petkau et al.,2021).
Although ALS and FTD are considered different, distinct diseases (ALS primarily affects motor function, while FTD is characterized by cognitive/social impairment), there is robust evidence of overlapping symptoms, as well as common pathological and genetic hallmarks. They are currently considered part of a clinic-pathological spectrum rather than two completely independent diseases. This is further supported by the discovery of a mutation in the C9ORF72 gene which can cause both diseases. The expansion of hundreds of repeats of the GGGGCC hexanucleotide is the most frequent mutation related to the development of ALS and FTD (DeJesus-Hernandez et al., 2011; Renton et al., 2011).This represents approximately 25% of familial FTD, 6% of sporadic FTD and 40% of familial ALS (Couratier et al., 2017).The C9ORF72 protein is highly abundant in the brain and spinal cord, where it can be found in the presynapsis, nucleus and cytoplasm of neurons and glia (Frick et al., 2018). Frontal cortices of patients with C9ORF72 mutation show significantly reduced levels of its transcript but a modest decrease of protein levels (Saberi et al., 2018; Braems et al., 2020).Normal C9ORF72 protein participates in autophagy, vesicle trafficking, and clearing of aggregated proteins; importantly,C9ORF72 mutations impair SG assembly and cause cellular hypersensitivity to stress signals (Maharjan et al., 2017).
?t is noteworthy that the affected proteins in ALS and/or FTD can be grouped into a few different functional categories.These include regulation of RNA metabolism (TDP-43,C9ORF72, FUS, hnRNPA1, ataxin 2, senataxin), antioxidant defense (SOD1), intracellular trafficking (optineurin, VAPB),modulation of cytoskeleton dynamics (PFN, tau), protein degradation regulation (VCP, ubiquilin 2, SQSTM1) and inflammation (progranulin) (reviewed in Ghasemi and Brown,2018).
Despite the fact that the full molecular mechanisms of these disorders are unknown, there is strong evidence that the disruption of protein homeostasis systems contributes to the progression of these neurodegenerative diseases.
The Link between TDP-43 and Translation in Normal and Pathological Situations
Physiological roles of TDP-43
Most sporadic and familial cases of ALS and approximately half of the cases of FTD present ubiquitinated protein inclusions that are positive for TDP-43 (Neumann et al., 2006).Interestingly, aggregates containing either wild-type TDP-43 or mutated versions of the protein have been associated with the development of ALS and FTD. Mislocalization of TDP-43 to the cytoplasm, its incorporation into aggregates or inhibition of the proteasome can lead to protein malfunction.Liquid-liquid phase interactions, nucleocytoplasmic shuttling,proper folding, and self-regulation of expression are key factors that ensure normal TDP-43 function. Protein shuttling usually depends on nuclear localization (NLS) and nuclear,exporting signal amino acid sequences. Although most ALSrelated mutations have been identified within the glycinerich C-terminal region, some models that express TDP-43 with mutated NLS recapitulate key ALS features. For example, our inducible TDP-43-?NLS mouse model display neurodegeneration, gliosis, changes in gene expression,motor, social and cognitive abnormalities (Igaz et al., 2011;Alfieri et al., 2014). Alterations in TDP-43 sequence led to aggregation, changes in solubility, sequestration of other TDP-43 molecules and seeding in a prion-like fashion, and these events have been shown to induce neurodegeneration. As mentioned before, TDP-43 proteins that harbor ALS-linked mutations in the C-terminal region (i.e., A315T and G335D)can enhance self-aggregation and cross-seeding (Guo et al.,2011; Jiang et al., 2016). ?n general, mutations in this region increase its aggregation propensity in a prion-like fashion,promoting fibril formation that leads to toxicity-mediated cell death (Guo et al., 2011; Smethurst et al., 2016).
Modulation of protein synthesis by TDP-43
TDP-43 is an RNA-binding protein that can directly or indirectly participate in protein synthesis. This includes functions such as modulation of mRNA metabolism by interacting with regions of specific mRNAs, regulating their trafficking, stability, accessibility, and translation. Nagano et al. (2020) have shown in axons of cortical neurons that mRNAs encoding ribosomal proteins are transported by TDP-43 (bound through its 5′-UTR region), and that this trafficking is necessary for normal ribosomal assembly and functionality. Also, physiological TDP-43 represses translation of transcripts involved in spinogenesis, synaptic plasticity,and neurodevelopment through binding to 3′-UTR regions at specific sequences, sometimes acting as an adaptor protein(Majumder et al., 2016). This repression can decrease at certain developmental stages and facilitate spinogenesis and neuronal maturation during brain development (Majumder et al., 2012). ?t is interesting to note that TDP-43 also has a role in mRNA stability, indicated by reduced levels of transcripts involved in ribosomal biogenesis in ALS patient-derived cells and tissue samples (Tank et al., 2018). These mRNAs were particularly rich in motifs that are recognized by RNA binding proteins. Importantly, TDP-43 controls its own homeostasis through binding to its transcript and downregulating its levels(Ayala et al., 2011).
Dysregulation of TDP-43 expression by diminished degradation, mislocalization, reduced or increased expression or by genomic knock-down can have drastic consequences for neurons. For example, overexpression of an ALS-associated TDP-43 mutant in drosophila leads to sequestration of mRNAs encoding chaperone Hsc70-4/HSPA8 away from ribosomes,resulting in impaired endocytosis of synaptic vesicles at the neuromuscular junction (Coyne et al., 2017). Interestingly,Russo et al. (2017) showed in vitro that cytoplasmic localization of TDP-43 can inhibit global protein synthesis by interacting directly with the ribosomal protein RACK1,which can promote the formation of aberrant inclusions.More recently, we demonstrated in vivo (using two different methods, SUnSET in brain slices and polysome profiling) that expression of TDP-43-?NLS decreases global brain translation in a transgenic mouse model (Charif et al., 2020).
During exposure to different stresses, TDP-43 is recruited into SG, which are membrane-less cytoplasmic RNA granules that sequester non-essential mRNAs and transcription factors.This represses their translation until the insult (i.e., altered proteostasis) is resolved, leading to SG disassembly (Fernandes et al., 2018). Protein-protein and protein-RNA interactions drive the reversible assembly. Some of the proteins that integrate SG have RNA-binding domains and a prion-like,intrinsically disordered domain within its sequence that is particularly prone to aggregation. TDP-43 plays a key role in SG formation and disassembly, as loss of TDP-43 decreases SG formation (McDonald et al., 2011). ?n addition, it has been shown that disease-linked mutations of TDP-43 can enhance SG formation (Liu-Yesucevitz et al., 2010). This latter work showed that inclusions of both wild-type and mutated TDP-43 can be disrupted by translational inhibitors that affect or impair SG assembly.
Additional TDP-43-related pathological mechanisms
Pathological propagation of TDP-43 proteinopathy can be achieved by the seeding of other TDP-43 proteins or exosomemediated, cell-to-cell dissemination. These processes are not mutually exclusive. For example, it has been demonstrated that aberrant TDP-43 (i.e., insoluble, hyperphosphorylated)can act as a template to modify normal TDP-43, inducing its aggregation and the formation of protein inclusions in vitro(Smethurst et al., 2016). Importantly, injection of insoluble TDP-43 from ALS or FTLD-TDP brains resulted in speeddependent aggregation and transmission to surrounding cells(Nonaka et al., 2013). Porta et al. (2018) have reproduced these results both in vivo (mouse model expressing TDP-43 with mutated NLS sequence) and in vitro (doxycyclineinducible wild-type and mutated NLS cell line). In the latter study, remarkably, seeding effects were more pronounced if TDP-43 was mislocalized to the cytoplasm.
Modulation of Translation as a Therapeutic Approach in Neurodegenerative Diseases
Normal brain function entails changes in synapses (for example, new synapse formation or strengthening of established synapses), and these processes heavily rely on rapid protein synthesis. This can be achieved de novo or by translating pre-localized mRNAs in synaptic terminals,ensuring quick availability of synaptic proteins (Vlatkovic and Schuman, 2016). Altered translation of mRNAs critical for synaptic development, plasticity and memory formation,or of mRNAs encoding translation-associated proteins can lead to neurodegenerative diseases or be a sign of them taking place. For instance, AD patients show reduced levels of proteins involved in translation, particularly in brain areas associated with memory processing and behavior (Garcia-Esparcia et al., 2017). Among these proteins, several initiation and elongation translation factors were affected. Perhaps the most relevant elongation factor controlling protein synthesis is e?F2α because it acts as a negative regulator of translation.Protein misfolding, mislocalization and accumulation can trigger cellular responses leading to e?F2α phosphorylation and subsequently to translational repression. If prolonged,this event can cause neuronal death.
As mentioned, high levels of abnormally folded or unfolded proteins trigger the UPR, which implies the activation of ER sensors. In particular, the ER sensor PERK phosphorylates e?F2α, resulting in global translation inhibition (Uppala et al., 2018). Altered levels of e?F2α and other cellular stress markers have been detected in samples from ALS patients and animal models of this disease (?lieva et al., 2007). Modulating the phosphorylation of e?F2α or upstream factors in this pathway seems to be an attractive target for treatment.e?F2α is phosphorylated by e?F2B (a guanine nucleotide exchanger), while phospho-e?F2α acts as an inhibitor of eIF2B (Krishnamoorthy et al., 2001). Interestingly, loss of function mutations in all e?F2B subunits have been linked to an autosomal recessive disorder termed leukoencephalopathy with vanishing white matter (van der Knaap et al., 2002).Key features of this disease include progressive neurological symptoms and myelin loss. The responses that tend to restore proteostasis (i.e., integrated stress response or ISR) have been shown to be exacerbated in mice models that recapitulate the disease. Pharmacological intervention with 2BAct (an e?F2B activator) can normalize the proteome without affecting this protein’s levels. As a result, the signs of the disease are halted (Wong et al., 2019). In line with this, another promising drug is ISRIB (acronym for Integrated Stress Response Inhibitor), a small molecule that blocks the PERK branch of the UPR. This downstream modulation of the effects of e?F2α phosphorylation has shown cognitive enhancement and many other benefits associated with proteostasis restoration in cellular and animal models of proteostatic stress (Sidrauski et al., 2013; 2015a) and brain injury (Chou et al., 2017). Very recently, Oliveira et al. (2021) showed that attenuation of the ISR with ISRIB rescued hippocampal protein synthesis and, remarkably, corrected impaired synaptic plasticity and memory in mouse models of AD. A key advantage of ISRIB is that it confers neuroprotection without pancreatic toxicity, unlike the PERK inhibitor GSK2606414 (Halliday et al., 2015; Mercado et al., 2018). ?t is worth highlighting that GSK2606414 has been shown to mitigate TDP-43 toxicity both in flies and in primary neurons studies, resulting in reduced p-e?F2α levels and motor ability recovery (Kim et al., 2014).Interestingly, this compound reversed cognitive deficits and abrogated the development of prion disease in a mouse model, acting independently of prion protein propagation and accumulation (Moreno et al., 2012). Another example is guanabenz, an antihypertensive drug that blocks the stress-induced PP1 phosphatase regulatory unit PPP1R15A(also known as GADD34), but spares the constitutively expressed subunit PPP1R15B (known as CReP), promoting e?F2α phosphorylation without lethal effects (Tsaytler et al.,2011). This drug also inhibits the protein-folding activity of the ribosome in an e?F2α-independent fashion (Tribouillard-Tanvier et al., 2008). Guanabenz has shown neuroprotective effects in PD models (Sun et al., 2018). However, results are mixed when applied to ALS models, because it can produce amelioration or acceleration of the disease, and has no effects on lifespan or after disease onset (Wang et al., 2014; Vieira et al., 2015). As guanabenz, its derivative sephin1 showed similar selective inhibition of GADD34, with the benefit of lacking α2-adrenergic agonistic activity. ?ts administration prevented motor deficits, motor neuron loss and molecular alterations in mutant SOD1 mice (Das et al., 2015). A compound termed salubrinal, which blocks both GADD34 and CReP, has shown to be neuroprotective after traumatic brain injury (Wang et al.,2019) and attenuated the activation of all three UPR branches after rotenone treatment in neuro2a cells (Gupta et al., 2019).However, salubrinal treatment resulted in early neuronal loss and disease acceleration in prion-infected mice (Moreno et al., 2012). A summary of relevant examples of modulators of protein synthesis used to treat neurodegenerative disease models is shown inTable 1.
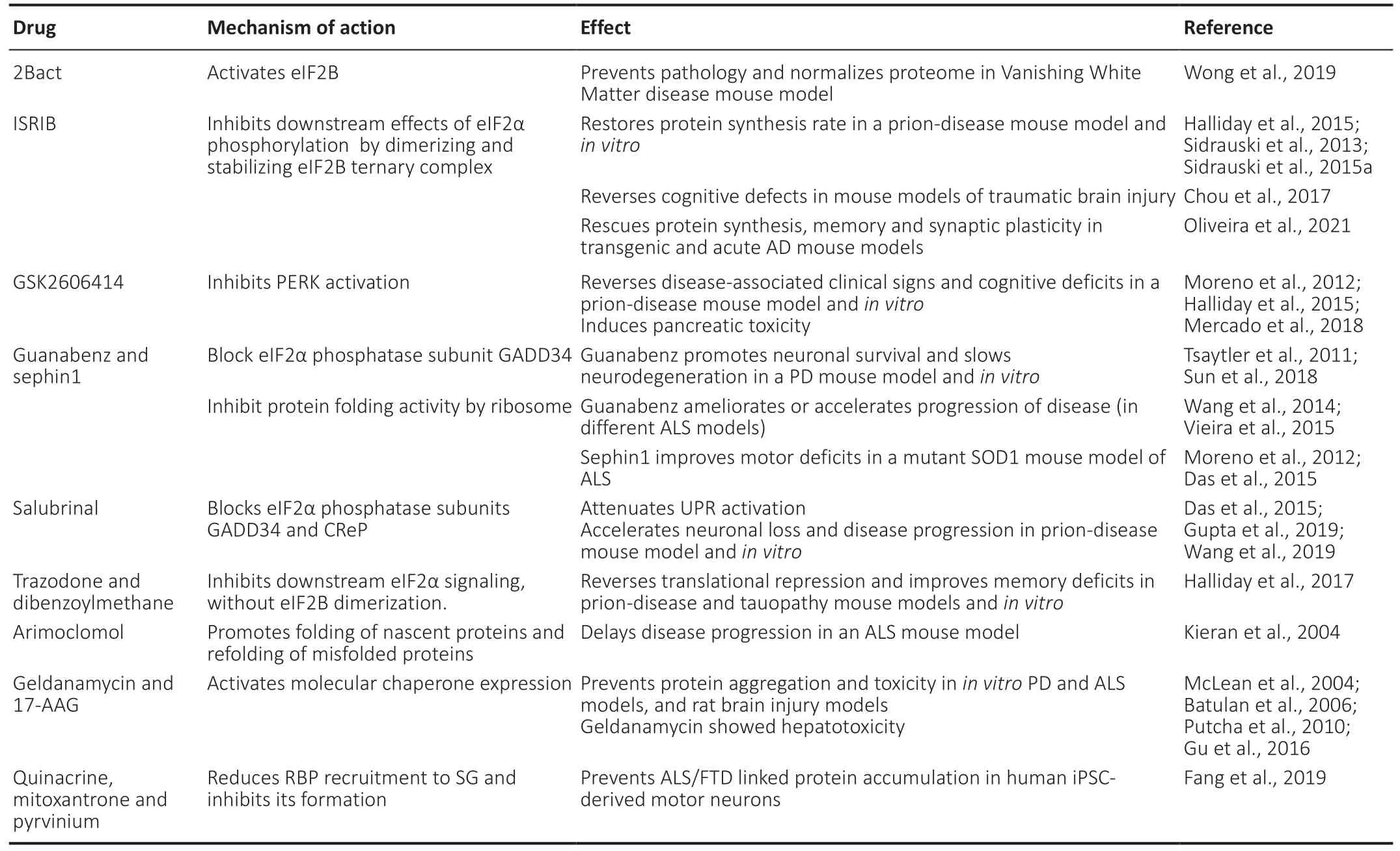
Table 1 |Examples of therapeutic drugs that aim to restore proteostasis in neurodegenerative diseases models
In a study published in 2017, Halliday et al. screened for safe therapeutic compounds targeting e?F2α phosphorylation effects. Trazodone hydrochloride (which is a licensed antidepressant) and dibenzoylmethane reversed translational repression triggered by p-e?F2α. This rescued behavioral and memory deficits and exerted a neuroprotective action bothin vitroand in prion disease and FTD-tau mouse models, without lowering p-e?F2α levels (Halliday et al., 2017). ?t is noteworthy that they act, in part, through a different mechanism than ?SR?B as they do not cause e?F2B dimerization (Sidrauski et al.,2015b; Halliday et al., 2017). Therefore, a promising and novel approach would be screening for and developing non-toxic therapeutic agents that restore protein synthesis rates and reestablish the normal activity of the UPR and ?SR.Proteostasis restoration can also be achieved with pharmacological chaperones. These small molecules bind and stabilize the target protein in the ER, allowing it to undergo the normal trafficking process to the Golgi apparatus and to find its final destination. Chaperones are very specific, targeting lysosomal enzymes, GPCRs, ion channels, transporters, and aggregation-prone proteins (reviewed in Tao and Conn, 2018).Also, a drug intervention aiming at modulating chaperone protein expression with arimoclomol has been relatively useful in delaying ALS progression in a mouse model, with a modest increase in lifespan and enhancement of the UPR (Kieran et al., 2004). However, a phase II/III trial of arimoclomol in SOD1-positive familial ALS patients showed only good tolerance and data consistency but no significant therapeutic effects (Benatar et al., 2018).
Geldanamycin is an antibiotic that inhibits the heat shock protein Hsp90, thus increasing activation of the heat shock transcription factor HSF-1 and the production of other chaperone molecules such as Hsp70 and Hsp40 (Sittler et al., 2001). Treatment with geldanamycin reduced protein aggregation-induced toxicity in models of neurodegenerative diseases. Examples includein vitroactivation of molecular chaperone expression and suppression of mutated huntingtin,α-synuclein, and SOD1 toxicity, as well asin vivoprotection against dopaminergic neurotoxicity in a fly PD model(McLean et al., 2004; Batulan et al., 2006). ?ts semi-synthetic analogue 17-AAG is blood-brain permeable and less toxic, and showed similar results in H4 neuroglioma cells transfected with α-synuclein (Putcha et al., 2010). Remarkably, 17-AAG exerted neuroprotection in a rat brain injury model,with the improvement of motor deficits (Gu et al., 2016). It also attenuated autophagic death of hippocampal CA1 cells and improved memory and learning functions after global cerebral ischemia induction in rats (Li et al., 2015). Another interesting approach is to find therapeutic drugs that target TDP-43 recruitment into SGs. Fang et al. (2019) screened for small molecules that may modulate SG biology. They found that a group of molecules with planar side chains (quinacrine,mitoxantrone, and pyrvinium) reduced SG formation and localization of TDP-43 in SG. Also, human induced pluripotent stem cell-derived motor neurons carrying ALS-linked mutations exhibited less TDP-43 accumulation in puncta when treated with these compounds.
Conclusions
Maintaining proper protein levels in neurons is vital, as several incurable neurodegenerative diseases are affected by altered amounts or post-translational modifications of specific proteins. Translational modulation aiming at restoring proteostasis in neurodegenerative disease seems to be a promising but poorly explored field with great potential.Studies on ALS/FTDin vitroandin vivomodels tackling this approach are relatively scarce. Some progress has been made in recent years, but the challenge is finding therapeutic compounds that are safe for humans, do not affect other vital cellular pathways and processes, and do not inadequately inhibit or over-activate the intrinsic stress response machinery.Screening for biologically active drugs is a suitable starting point, allowing for the discovery and development of several compounds that differ in their mechanisms of action and molecular structure. This is important in terms of formulation,administration and, what is crucial, time of action.Additionally, more focus should be put on substances that can disassemble pathological protein aggregates or, at least, that reduce their quantity or deleterious effects when assembled.In line with this, avoiding and preventing toxicity of protein aggregates is a key goal, especially in the early or mid-stages of neurodegenerative diseases. Based on the current evidence presented here, it is likely that keeping neuronal protein levels physiologically stable and avoiding its alteration could prevent further neurodegeneration, ameliorate symptoms, and greatly delay disease onset, allowing patients to improve their quality of life and overall life expectancy.
Acknowledgments:The authors want to thank Pedro Bekinschtein(INCYT, CONICET-Fundación INECO-Universidad Favaloro, Argentina)and Fernando Guerrieri (IRBI, Université de Tours, France) for valuable discussion and comments.
Author contributions:Review concept and design: SEC and LMI;manuscript preparation: SEC, MFV, LS and LMI; manuscript revision: SEC,MFV and LMI; literature search: SEC, MFV and LS; funding acquisition and project supervision: LMI. All authors approved the final version of the manuscript.
Conflicts of interest:The authors declare no conflicts of interest.
Financial support:This work was supported by research grants to LMI from University of Buenos Aires (UBACyT) and the Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT) under grants PICT 2015-0975 and PICT 2017-2140.
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix,tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.

- 中國神經(jīng)再生研究(英文版)的其它文章
- Rethinking the necessity of low glucose intervention for cerebral ischemia/reperfusion injury
- Microglia activation, classification and microgliamediated neuroinflammatory modulators in subarachnoid hemorrhage
- MicroRNA biomarkers in frontotemporal dementia and to distinguish from Alzheimer’s disease and amyotrophic lateral sclerosis
- A novel viewpoint in glaucoma therapeutics: enriched environment
- Neuronal reprogramming in treating spinal cord injury
- The role of L-arginine metabolism in neurocritical care patients