
Atomic Cu Sites Engineering Enables Efficient CO2 Electroreduction to Methane with High CH4/C2H4 Ratio
2023-11-18 09:45:18MinhanLiFangzhouZhangMinKuangYuanyuanMaTingLiaoZiqiSunWeiLuoWanJiangJianpingYang
Nano-Micro Letters 2023年12期
Minhan Li, Fangzhou Zhang, Min Kuang, Yuanyuan Ma?, Ting Liao, Ziqi Sun,Wei Luo, Wan Jiang, Jianping Yang?
ABSTRACT Electrochemical reduction of CO2 into high-value hydrocarbons and alcohols by using Cu-based catalysts is a promising and attractive technology for CO2 capture and utilization, resulting from their high catalytic activity and selectivity.The mobility and accessibility of active sites in Cubased catalysts significantly hinder the development of efficient Cu-based catalysts for CO2 electrochemical reduction reaction (CO2RR).Herein, a facile and effective strategy is developed to engineer accessible and structural stable Cu sites by incorporating single atomic Cu into the nitrogen cavities of the host graphitic carbon nitride (g-C3N4) as the active sites for CO2-to-CH4 conversion in CO2RR.By regulating the coordination and density of Cu sites in g-C3N4, an optimal catalyst corresponding to a one Cu atom in one nitrogen cavity reaches the highest CH4 Faraday efficiency of 49.04% and produces the products with a high CH4/C2H4 ratio over 9.This work provides the first experimental study on g-C3N4-supported single Cu atom catalyst for efficient CH4 production from CO2RR and suggests a principle in designing highly stable and selective high-efficiency Cu-based catalysts for CO2RR by engineering Cu active sites in 2D materials with porous crystal structures.
KEYWORDS CO2RR; Cu single-atom catalyst; g-C3N4; Methane; CH4/C2H4 ratio
1 Introduction
Cu-based electrocatalysts have received extensive attentions for CO2electrochemical reduction reaction (CO2RR),because of their unique ability to convert CO2into highvalue deep reduction products [1-5].Motivated by the great CO2-to-CO performance in CO2RR obtained on various carbon-supported single-atom catalysts, such as Ni [6-8],Fe [9, 10], Co [11, 12], and Zn [13], the Cu single-atom catalysts (Cu-SACs) have also been investigated for CO2RR [14].Similar to the nanostructured Cu-based materials [15], the reported Cu-SACs also exhibited selectivity characteristics toward various reduction products in CO2RR, including CO[16], CH4[17, 18], CH3OH [19, 20], C2H5OH [21, 22], and CH3COCH3(acetone) [23].The isolated Cu single atoms are usually regard as the main active centers for CO2RR,and the coordination environment and density of the atomic Cu species play a key role in the selectivity of the Cu-SACs.Cai and co-workers reported that carbon dots-supported Cu-SACs with unique Cu-N2O2sites enabled efficient CO2RR to CH4[24].Xu and co-workers found that O atom coordinated single Cu atoms supported on carbon underwent a reversible transition between atomic sites and Cunclusters under CO2RR condition, and the later catalyzed CO2into ethanol [21].Zheng and co-workers reported that the distance between the atomic Cu-Nxsites was critical for the selectivity between CH4and C2H4, where the neighboring Cu - Nxsites promoted the C-C coupling and decreased the CH4/C2H4ratio [25].During the synthesis of Cu-SACs, however, a high-temperature pyrolysis process is often involved,resulting in poorly defined coordination structure and the aggregation of the atomic Cu sites and thus poorer catalytic selectivity and less accessibility of the active sites [15, 26].Compared to the pyrolytic SACs, molecular catalysts with explicit and tunable structures are more preferred, due to their well-defined and uniform coordination environments[24].
The challenges in preparing more structural stable and well-configured Cu-SACs triggered our interest in searching more suitable catalyst supports to anchor the active single atoms in a stable and well-coordinated environment.Similar to the well-documented graphene single-atom support,graphitic carbon nitride (g-C3N4) has a perfect 2D morphology with single atomic thickness to ensure the best accessibility of the active sites.But differently, g-C3N4contains periodic heptazine units and well-defined “nitrogen pots”that consist of six lone pairs from pyridine-like nitrogen atoms among the heptazine units (nitrogen cavities) [27, 28].The abundant and periodic nitrogen cavities in the g-C3N4framework makes them become perfect anchoring points for incorporating dense metal atoms with defined distance and coordination numbers [29-31].Various metal-doped g-C3N4catalysts have been investigated for different electrocatalysis processes, such as hydrogen evolution reaction (HER) [32],oxygen evolution and reduction reaction (OER and ORR)[27, 33], nitrogen reduction reaction [34], and even CO2RR[35].Specifically, it has been reported that a Cu-C3N4catalyst exhibited strong CO2affinity for promoted CO2adsorption toward deep reduction for converting CO2into highvalue hydrocarbons.However, the Faraday efficiencies (FEs)of these deep reduction reactions were very low, and the role of the Cu sites incorporated in g-C3N4framework for CO2RR remains unclear [36].Therefore, further efforts in further enhancing the FEs of the Cu-g-C3N4catalytic system together with a clear mechanism understanding are urgently needed.
In this work, a facile thermal polymerization method is employed to synthesize Cu single atoms loaded in g-C3N4catalysts with different site distance and coordination environments (denoted as Cux-CN) for CO2RR by adjusting the ratios of Cu to g-C3N4in the precursors during the preparation.The synthesis-structure-activity analysis of Cux-CN catalysts demonstrated that the Cu single atoms anchored in the nitrogen cavities of g-C3N4are highly active and selective for the production of CH4.For the Cu0.05-CN catalyst with a coordination of N atoms in nitrogen cavities and a chemical state of dominant Cu+, the most optimal Cu-CN catalyst, presented the highest activity and CH4FE (49.04%)and reached 7.97 mA cm-2at -1.2 VRHEwith a high CH4/C2H4ratio of 9, which, to the best knowledge of authors,is the best CO2-to-CH4performance on g-C3N4-supported catalysts for CO2RR.Combining experimental evidence and density functional theory (DFT) calculations, it is revealed that one Cu single atom in one nitrogen cavity and coordinated with 4 N atoms of g-C3N4exhibit a preferred CH4catalytic pathway than the C2H4pathway, while the preferred pathway is reversed after forming an adjacent O-coordinated Cu atom.The coordination environment regulated Cux-CN catalysts for high-efficiency CH4production provide a feasible way to fabricate single-atom Cu active sites for efficient and selective CO2RR catalysis and offer some insights into the design of future electrocatalysts.
2 Experimental Section
2.1 Materials
Dicyandiamide (DCDA), copper (II) acetylacetonate(Cu(acac)2), Iron (III) acetylacetonate (Fe(acac)3), and nickel (II) acetylacetonate (Ni(acac)2) were purchased from Sinopharm Chemical Reagent Co., LTD.Potassium bicarbonate (ACS, 99.7-100.5%) and deuterlum oxide (D2O, 99.9 atom% D) were purchased from Shanghai Aladdin Biochemical Technology Co., Ltd.Nafion solution (5 wt% in mixture of lower aliphatic alcohols and water) was purchased from Sigma-Aldrich.Nafion 117 membrane was purchased from Shanghai Hesen Electric Co., LTD.Standard liquid products: methanol (> 99.9%), sodium formate (99.99%),ethanol (> 99.8% for GC), and n-propanol (99.99%) were purchased from Shanghai Macklin Biochemical Co., Ltd.Deionized water (18.2 MΩ cm-2) was used in this work.The gas products, including H2, CO, CH4, and C2H4, were calibrated using standard mixed gases purchased from Dalian Special Gases Co., LTD.
2.2 Preparation of Catalysts
To synthesis Cux-CN catalysts, 1 g of DCDA and certain amount of Cu(acac)2were well mixed in a mortar with the mass ratio of Cu(acac)2to DCDA of x (x = 0.01, 0.05, 0.2,and 0.5 in this work).The well-mixed and grinded powder in a quartz boat was placed in the middle of a tubular furnace.Then the calcination process starts from room temperature to 550 ℃ at 5 ℃ min-1and holds at 550 ℃ for 4 h in Ar atmosphere.After cooling to room temperature, the product was grinded into fine powder.CN sample was prepared by the same procedure except for the absence of Cu(acac)2.Nix-CN and Fex-CN materials were synthesized by the same method using Ni(acac)2and Fe(acac)3as the metal sources.
In order to leach out the Cu in Cu0.05-CN catalyst for comparison, the Cu0.05-CN catalyst was washed with 1.0 M nitric acid for 12 h at room temperature.After washing with deionized water to neutral pH, the sample that labeled as 0.05-CN was obtained, in which almost all of the Cu metal was removed by acid treatment (Table S1).
2.3 Characterization of Materials
Transmission electron microscope (TEM) images were acquired using JEOL 2100F operated at 200 kV.High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and elemental mapping analysis were performed on Talos F200S operated at 200 kV.Powder X-ray diffraction (XRD) was obtained on Bruker D2 Phaser with a 2θ ranging from 10° to 90° using a Cu Ka X-ray.X-ray photoelectron spectroscopy (XPS) measurements were taken on Escalab 250Xi device.1H nuclear magnetic resonance (NMR) spectra were collected on Bruker AVANCE III 600 MHz nuclear magnetic resonance spectrometer.Solidstate13C magic angle spinning nuclear magnetic resonance(MAS NMR) spectra were acquired with a Bruker AVIII400 spectrometer with a 4 mm MAS BB-1H probe at frequency of 100.63 MHz.Elemental analysis (EA) was performed with Elmentar Vario EL III elemental analyzer to quantitatively determine the elemental content of C, H, and N.Fourier transform infrared (FTIR) spectrum was measured with a Nicolet 6700 FTIR spectrophotometer in the range of 400-4000 cm-1using KBr pellet technique.Spherical aberration-corrected transmission electron microscope(AC-TEM) was performed on Themis ETEM (Thermo Fisher Scientific) transmission electron microscope.The measurements of X-ray absorption spectroscopy (XAS) at the Cu K-edge containing the X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) were taken at the beamline BL14W1 of Shanghai Synchrotron Radiation Facility (SSRF), China.The data processing of XAS measurements was performed using the Demeter software package.Nitrogen sorption-desorption measurements were taken at Micromeritics ASAP2046 machine.Before the measurements, the samples were degassed in a vacuum at 180 °C for at least 10 h.The Brunauer-Emmett-Teller (BET) method was utilized to calculate the specific surface areas using the adsorption data at P/P0= 0.02-0.20.The pore size distribution was calculated from the adsorption branch by using the Barrett-Joyner-Halenda (BJH) model.
Electrochemical in situ attenuated total reflection Fourier transform infrared (ATR-FTIR) reflection spectroscopy was investigated on a Fourier transform infrared spectrometer(FTIR, Nicolet iS50, Thermo Fisher Scientific) equipped with a liquid nitrogen-cooled mercury cadmium tellurid (MCT)detector.The measurement was conducted in a homemade electrochemical cell equipped with a Pt-mesh and an Ag/AgCl as counter and reference electrodes, respectively.An Au-coated Si crystal loaded with catalysts was embed into the bottom of the cell to serve as the working electrode.CO2-saturated 0.1 M KHCO3is used as the electrolyte for the in situ ATR-FTIR measurement, during which CO2gas is continuously bubbled.Chronoamperometry is used for the in situ CO2RR test, and the spectrum is collected by 32 scans with 4 cm-1resolution.All spectra were subtracted with the background.
2.4 Electrode Preparation
Cux-CN catalyst (1 mg) and carbon black (0.25 mg) were dispersed in methanol solution (190 uL).Subsequently, 10 uL of Nafion (5 wt%) was added, followed by ultrasonication for at least 1 h.Then 6 uL catalyst ink was dropped onto a L-type glass carbon electrode with a diameter of 4 mm (geometric area: ~ 0.1256 cm2) using a pipette and dried under ambient air.The catalyst loading was about 0.24 mg cm-2.The catalysts were electrochemically activated in CO2-saturated 0.1 M KHCO3solution by CV (10 scans) from - 0.5 to -1.5 VRHE.
2.5 Electrochemical Reduction of CO2
CO2electrolysis was carried out in a gastight, custom-made two-compartment cell, in which the working electrode was separated from the counter electrode by a Nafion 117 membrane.Ag/AgCl (saturated KCl) was used as the reference electrode, and the three-electrode setup was connected to a potentiostat (Biologic VMP3).A 0.1 M KHCO3electrolyte solution was used for all CO2RR tests, and the electrolyte in the cathodic compartment was stirred at a constant rate of ~ 300 rpm during electrolysis.Before CO2RR, the catholyte was bubbled with CO2(99.99%) for 30 min to reach saturation, and CO2was kept purging into the cathodic compartment at 10 sccm with stirring at ~ 500 rpm during the CO2RR.To determine the Faradaic efficiencies (FEs) of the reduced products, chronoamperometry was performed for 1 h at constant iR-corrected potentials.For the long-term stability test, the CO2-saturated 0.1 M KHCO3electrolyte was extracted and replaced every 4 h to determine the liquid products.The FEs of gas products were calculated and recorded when the FEs were stable during the last 3 h.
All potentials were measured against an Ag/AgCl reference electrode (3.5 M KCl, stored in a 3.5 M KCl solution before and after use) and converted to the reversible hydrogen electrode (RHE) scale by
The resistance between the reference and working electrodes was measured by potential electrochemical impedance spectroscopy (PEIS) and the ohmic drop was compensated automatically by the software (80%) and the rest 20% remained uncompensated.All the potentials in the text were iR-corrected if not otherwise specified.Generally, the value of resistance(R) was about 110-115 Ω in CO2-saturated 0.1 M KHCO3.
2.6 Analysis of Gas and Liquid Products
Gas products from the cathodic compartment during CO2RR were analyzed using a GC-2014 (Shimadzu) equipped with a TCD detector and two FID detectors, one of which was coupled with a methanizer to detect CO concentration.Highpurity Ar (99.999%) was used as the carrier gas.The calibration curves of gas and liquid products can be referred to our previous publication [3].
The Faradaic efficiencies of the gas products were calculated by the GC data using the following equation:
where v is gas flow rate measured by a flowmeter (Beijing FLOWS instruments Co, Ltd), which is 10 sccm for all the tests,ythe measured volumetric content of the gas product,n the number of electrons required to form the gas products,and n = 2, 2, 8, and 12 for H2, CO, CH4, and C2H4, respectively, F the Faraday constant (96,485 C mol-1), andjthe average current density.
Liquid products were analyzed by a 600 MHz NMR spectrometer (Bruker Avance 3 HD 600 MHz) using a presaturation technique to suppress water peak.To take1H NMR measurement, 800 uL of electrolyte sampled after CA or stability tests was mixed with 100 uL DMSO standard solution (100 ppm) and 100 uL D2O.The faradaic efficiencies of liquid products were calculated as follows:
where nlis the total content of certain liquid products in the catholyte, which was calculated by the concentration and the volume of the catholyte (45 mL), and n is the number of electrons required to form the liquid products, and n = 2,6, 8, 12, and 18 for formate, methanol, acetate, ethanol, and 1-propanol, respectively.
2.7 Computational Details
We carried out all the DFT calculations in the Vienna ab initio simulation (VASP5.4.4) code [37].The exchange-correlation is simulated with PBE functional, and the ion-electron interactions were described by the PAW method [38, 39].The vdWs interaction was included by using empirical DFTD3 method [40].The Monkhorst-Pack-grid-mesh-based Brillouin zone k-points are set as 3 × 3 × 1 for all periodic structure with the cutoffenergy of 450 eV.The convergence criteria are set as 0.025 eV A-1and 10-4eV in force and energy, respectively.A 20 ? vacuum layer along thezdirection is employed to avoid interlayer interference.
The free energy calculation of species adsorption (ΔG)is based on N?rskov et al.’s hydrogen electrode model [41].
Herein ΔE, ΔEZPE, and ΔS, respectively, represent the changes of electronic energy, zero-point energy, and entropy that caused by adsorption of intermediate.The entropy of H++ e-pair is approximately regarded as half of H2entropy in standard condition [42].
3 Results and Discussion
3.1 Synthesis and Characterizations of Cux-CN Catalysts
The single Cu atoms anchoring in the nitrogen cavities of g-C3N4catalysts with tailoring Cu coverage and coordination (Cux-CN) were synthesized via an in situ thermal polymerization method by using dicyandiamide (DCDA)and copper (II) acetylacetonate (Cu(acac)2) as precursors for g-C3N4host and Cu active species, respectively (Fig.1a).In the synthesis, Cu(acac)2and DCDA with a mass ratio of x (x = 0.01, 0.05, 0.2, and 0.5) in Cux-CN catalysts were mixed and then calcined under N2at 550 ℃ to obtain the Cux-CN catalysts.The pure g-C3N4(denoted as CN) is yellowish powders.With the increase in Cu contents in Cux-CN catalysts, the color gradually turns into dark brown (Fig.1a).
The morphology of the catalysts and the dispersion state of Cu species are first characterized.The TEM and highangle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images of both the CN and the Cux-CN catalysts (Figs.1b and S1-S2) confirm their stacking layered structures and exclude the existence of crystalline or aggregated Cu species in these catalysts.The stacked layers gradually became fragmentary with the increasing Cu content in Cux-CN catalysts, demonstrating the increasing surface area and pore structure.Thus, the as-prepared material is investigated by the N2adsorption-desorption measurements.As expected, after loading Cu the surface area and porosity of Cux-CN catalysts increase significantly as compared with the pristine CN sample (Fig.S3).As observed on the Cu0.05-CN catalyst by aberration-corrected HAADFSTEM, the bright dots scatteringly distribute in the g-C3N4host, indicating the presence of isolated Cu atoms in the catalyst (Fig.1c).Interestingly, even at high Cu contents,isolated Cu atoms appeared at a high density rather than forming crystalline species or aggregated clusters, for example, in the Cu0.5-CN catalysts (Fig.S2c), which is consistent with the TEM results.The uniform distribution of Cu, C,N, and O elements in the Cu0.05-CN catalyst is further verified by the elemental mappings (Figs.1d and S4).The high density of the atomic sites shortens the distance between single atoms, which may impact on both the activity and the selectivity of the catalysts [25, 43].Besides, this facile strategy can be extended to other transition metals to form g-C3N4-supported single metal atoms, such as Ni and Fe(Figs.S5-S6).
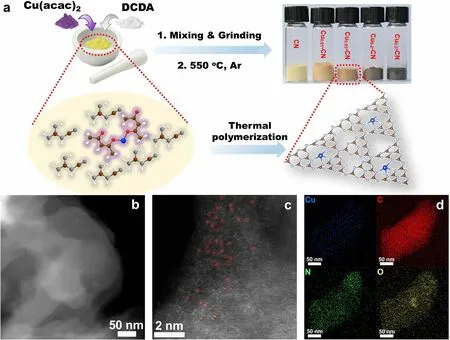
Fig.1 a Schematic illustration of the synthesis of Cux-CN catalysts and the optical photographs of the as-synthesized Cux-CN catalysts.b HAADF-STEM image, c aberration-corrected HAADF-STEM image, and d EDS mapping of Cu0.05-CN catalysts
The XRD patterns of the CN, the Cu0.01-CN, and the Cu0.05-CN catalysts exhibit the (100) and (002) peaks of g-C3N4appeared at 13.0° and 27.2°, corresponding to the repeating in-plane melem units and the stacking of graphite-like layer structure, respectively (Fig.2a) [28].For the Cu0.2-CN and Cu0.5-CN catalysts, the intensity of (002) peak decreased and the (100) peak disappeared, indicating the loss of periodical repeatability of the in-plane melem units in these two catalysts.No phase of Cu species appeared in all catalysts, which excludes the existence of crystalline Cu species and indicates the likely chemically coordination of Cu into the g-C3N4host in the form of Cu-Nystructure (y is the coordinated N number) [28].The FTIR spectra of all samples exhibit a broad peak between 3100 and 3600 cm-1that can be attributed to N-H and O-H stretching [44, 45],while the weak peaks appear at ~ 2176 cm-1can be assigned to the stretching of triple C≡N bond, which is considered as defects within g-C3N4[46].The increasing intensity of C≡N peaks support the fact that the defect increases with increasing Cu content in Cux-CN catalysts.The typical bands of the stretching vibrations of C-N heterocycles and breathing mode of s-triazine units appears in at ~ 1250-1635 and at ~ 810 cm-1, respectively [47-49].Notably, at low Cu loads, these two characteristic FTIR spectra of the Cu0.01-CN and Cu0.05-CN catalysts are almost identical with that of the pure g-C3N4(Fig.2b), indicating the well-preserved g-C3N4frameworks.On the contrary, these characteristic bands are significantly weakened in the Cu0.2-CN and Cu0.5-CN catalysts, indicating the high Cu loads in the catalysts can partially damage of the C-N heterocyclic structure [50].To further confirm the local structures of the Cux-CN catalysts,solid-state13C nuclear magnetic resonance (13C NMR) spectra were carried out on the Cux-CN with various Cu contents(Fig.2c).Two distinct peaks at chemical shift of 156.3 and 164.0 ppm, which are assigned to the C atoms connected to different N atoms in C-N heterocycle [49], are observed in the CN, Cu0.01-CN, and Cu0.05-CN samples, indicating the skeleton structure of g-C3N4are well retained and the introduced Cu atoms are likely to incorporate into the nitrogen cavities among C-N heterocyclic structures in these samples.However, the two peaks dramatically attenuate for the Cu0.2-CN and Cu0.5-CN catalysts, demonstrating the destruction of g-C3N4framework by increasing Cu(acac)2content in the preparation.Obviously, the XRD, FTIR, and13C NMR results explicitly suggest that the basic structure units and framework of the g-C3N4host are well retained for Cu0.01-CN and Cu0.05-CN catalysts with low Cu loads,which is likely associated with the preferential incorporation of isolated Cu atoms in the nitrogen cavities of g-C3N4with Cu-N structures.On the other hand, the destructed C-N heterocycles and g-C3N4framework of Cu0.2-CN and Cu0.5-CN catalysts indicate the changed atomic coordination at relatively high Cu loads.
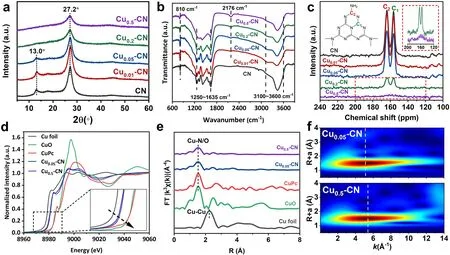
Fig.2 a XRD patterns of Cux-CN catalysts.b FTIR spectra of Cux-CN catalysts.c Solid-state 13C NMR spectra of Cux-CN catalysts.d Cu K-edge XANES spectra of Cu foil, Cu0.05-CN, Cu0.5-CN, CuPc, and CuO samples.e Fourier transform (FT) EXAFS spectra of Cu K-edge of Cu foil, Cu0.05-CN, Cu0.5-CN, CuPc, and CuO samples.f Wavelet transform (WT) of Cu0.05-CN and Cu0.5-CN catalysts
To get a clearer understanding on the chemical states and coordination of Cu atoms in the Cux-CN catalysts,XAFS measurements were taken together with Cu foil,copper (II) phthalocyanine (CuPc), and CuO as references.As illustrated by the X-ray absorption near-edge structure (XANES) in Fig.2d, the positions of Cu K-edge for the Cu0.05-CN and Cu0.5-CN catalysts are close, which are located between the Cu K-edge edges of Cu foil and CuO.The average oxidation states of Cu in the Cu0.05-CN and Cu0.5-CN catalysts are found to be close to + 1 by fitting (Fig.S7) [51].The Fourier transformed extended EXAFS spectra withk3-weight of Cu0.05-CN, Cu0.5-CN,and the references are depicted in Fig.2e.It is found that the Cu0.05-CN and Cu0.5-CN catalysts exhibited only one dominating peak around 1.53 ?, which could be assigned to the scattering of either Cu-N or Cu-O coordination[24].As references, CuO shows a main peak attributed to Cu-O at 1.53 ? while the Cu-Cu coordination in Cu foil is at 2.24 ?.These results further verify that no Cu-Cu interaction existed but only the isolated Cu atoms in the Cux-CN catalysts, even for Cu0.5-CN with high Cu loads.Notably, the Cu0.5-CN catalyst exhibited a lower intensity at 1.53 ? than that of the Cu0.05-CN catalyst, suggesting a possible different coordination environment achieved at higher Cu contents [52, 53].Due to the great resolution in both k and R spaces, the wavelet transform of Cu K-edge EXAFS spectra were performed to investigate the atomic configuration of Cu0.05-CN and Cu0.5-CN catalysts(Fig.2f).summits at 5.2 and 5.4 ?-1were identified for Cu0.05-CN and Cu0.5-CN, respectively, in the k space.Although the wavelet transform of Cu K-edge EXAFS is unable to discriminate the N and O coordination completely [24], these results confirm the coordination environment of the Cu single atoms in Cux-CN can be varied as the variation of Cu loading amounts.
The composition, valence state, and detailed structural and binding information of the Cux-CN catalysts were further investigated by various characterizations.To verify the actual Cu contents in each catalyst, inductively coupled plasma-atomic emission spectrometry (ICP-AES) was employed.It reveals that the actual Cu contents are proportional to the Cu(acac)2addition amounts, and the Cu0.05-CN catalyst had a Cu content of 0.954 wt% (Table S1).The compositions of the CN and Cux-CN catalysts were then probed by an elemental analysis (EA).As shown in Fig.3a, the CN and Cux-CN catalysts possess similar C content, while the N and H contents decrease distinctly for the Cu0.2-CN and Cu0.5-CN catalysts.Similarly, the significant decrease of N content in these two catalysts was also observed by the atomic percentage determined by XPS (Figs.3b and S8),accompanying with the increases in Cu and O contents.The composition variations indicate that the Cu atoms are successfully introduced in g-C3N4host accompanied by the introduction of O atoms, which is hypothesized to be caused by the use of oxygen-containing copper salt (copper acetylacetonate) in the polymerization process of g-C3N4.
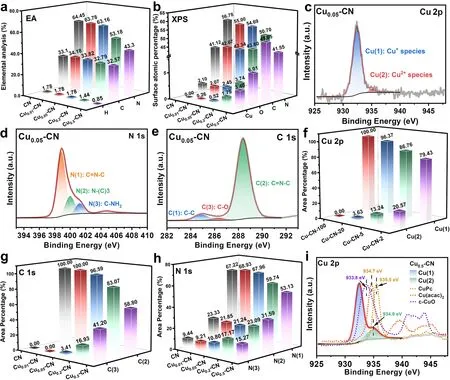
Fig.3 a Elemental analysis of the catalysts.b Surface composition of the catalysts determined by XPS.c-e Peak fitting of Cu 2p, N 1s, and C 1s spectra of the Cu0.05-CN catalyst.f-h Peak fitting summary of Cu 2p, C 1s, and N 1s spectra of the catalysts.i Comparison of Cu 2p spectra of the Cu0.5-CN catalyst with other cupric compounds
The bonding and chemical states of the elements in CN and Cux-CN catalysts are analyzed by the XPS spectra collected on the Cu, C, and N elements of all catalysts.The Cu 2ppeaks can be fitted with two peaks (Figs.3c and S9).The Cu(1) peak at about 932.4 eV is predominant in the Cu0.01-CN and Cu0.05-CN catalysts and can be ascribed to Cu0or Cu1+species, while the Cu(2) peak at about 935.0 eV ascribed to Cu2+species increases significantly in the Cu0.2-CN and Cu0.5-CN catalysts with higher Cu contents (Fig.S6d-e).Further, according to the XANES results and the Cu LMM Auger spectra of Cux-CN catalysts, it can be concluded that Cu(1) peaks in Cux-CN should be ascribed to Cu1+species, as the overwhelming Cu1+peaks and negligible Cu0peaks in the Cu LMM spectra (Fig.S9f) [54].The N 1sspectra can be deconvoluted into three states of N(1), N(2), and N(3) at about 398.7, 399.8, and 401.1 eV, respectively (Figs.3d and S10), as well as a small peak at 404.9 eV, which corresponds to the π-excitation.The N(1), N(2), and N(3)peaks can be attributed to thesp2hybridized nitrogen in C-N heterocycles (C = N-C), the tertiary N (N-(C)3), and the N in amino functional groups (C-NH2), respectively[55].The C 1speaks can also be fitted with three peaks denoted as C(1), C(2), and C(3) at about 284.8, 288.3,and 286.6 eV, respectively (Figs.3e and S11).The C(1)peak can be ascribed to graphitic carbon and used for calibration.The C(2) peak is the main component of C 1sspectra for all catalysts, which should be the C atoms in the C-N heterocycles (C = N-C).The C(3) state gradually emerges and rises with the increase of Cu contents in Cux-CN catalysts, which may attribute to the C in a C-O structure [55-57].
The fitting results of the Cu 2p, C 1s, and N 1sspectra are summarized in Fig.3f-h.In accordance with the structural characterization of the catalysts in Fig.2, the fitting results show that the species and ratio of C and N are almost unaffected for the Cu0.01-CN and Cu0.05-CN catalysts.Combined the results of XAFS measurements, it is reasonable that the isolated Cu atoms preferentially embed nitrogen cavities to form Cu-N coordination structures with little effects on the C-N heterocyclic structure in these two catalysts.However,with the increasing addition of Cu(acac)2in the preparation, the significantly reduced N(1) and C(2) peaks further confirm the destruction of the C-N heterocyclic structure of the g-C3N4host in the Cu0.2-CN and Cu0.5-CN catalysts.Meanwhile, the concurrent increases of the Cu(2) species and the O atomic ratio for the Cu0.2-CN and Cu0.5-CN catalysts imply the emerging Cu2+species is related to the increasing C-O structures, which is evidenced by increased C(3) peaks and the O 1sspectra (Figs.3g and S12).Further,by comparing the Cu 2pspectra of the Cu0.5-CN catalyst with some references, including commercial CuO powder,Cu(acac)2, and CuPc, it is found that the binding energy of Cu2+species in the Cu0.5-CN catalyst is close to Cu(acac)2(Fig.3i), indicating the structure of the emerging Cu2+sites in the Cu0.2-CN and Cu0.5-CN catalysts are likely the O-coordinated structure [58].Therefore, the possible evolution of the atomic configurations of the Cux-CN catalysts can be drawn from the above analysis.In the Cu0.01-CN and Cu0.05-CN catalysts, the atomic Cu sites predominantly embed in the nitrogen cavities of the g-C3N4host, forming Cu-N coordination without affecting its framework structure.However, in the Cu0.2-CN and Cu0.5-CN catalysts, the O-coordinated Cu species emerge substantially accompanied with the partial destruction of the C-N heterocycles and g-C3N4framework.
3.2 Catalytic Performance of CO2RR
The CO2RR performance of the CN and Cux-CN catalysts was evaluated in a H-cell with 0.1 M KHCO3as the electrolyte.The potential dependent FE of different products on the CN and Cux-CN catalysts was collected by a chronoamperometry test of 1 h in the potential range from -0.9 to -1.3 VRHE(Fig.4a).The gaseous products account for the major reduction products on all catalysts (Figs.4b and S13-S14).Compared with the pristine g-C3N4, the Cux-CN catalysts exhibited higher activity and selectivity toward CH4in the tested potential range.The highest FE of 49.04% at -1.2 VRHEfor CH4reached on the Cu0.05-CN catalyst with a CH4current density of 7.97 mA cm-2, which further increased to 9.78 mA cm-2at -1.3 VRHE(Fig.4c-d).Although the CN catalyst also exhibits certain activity toward CO2RR in the potential range, it only shows quite low CH4activity and selectivity with a maximum CH4current density of 2.11 mA cm-2at -1.3 VRHE.Clearly, the enhanced CH4production obtained on the Cux-C3N4catalysts indicates the incorporated Cu sites in these catalysts are active sites for CO2RR catalysis.Probably contributed by the stable configuration of single Cu atoms in the Cu0.05-CN catalysts(Fig.S15), the catalytic stability up to 10 h of continuous CO2RR test was performed on the Cu0.05-CN catalyst at -1.2 VRHE, which showed a slight decrease within the stability test (Fig.4e).
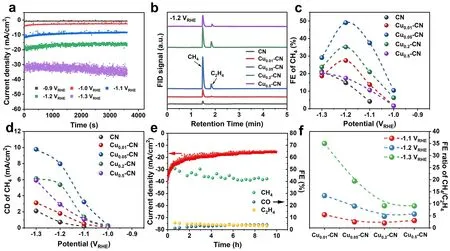
Fig.4 CO2RR performance of CN and Cux-CN catalysts.a Typical chronoamperometry curves at different potentials on Cu0.05-CN catalysts.b FID signals of GC showing the main gaseous CO2RR products of CH4 and C2H4 on Cux-CN catalysts.c FE of CH4 at different potentials on Cux-CN catalysts.d Current density (CD) of CH4 at different potentials on Cux-CN catalysts.e Catalytic stability of Cu0.05-CN catalyst during 10 h of CO2RR test at -1.2 VRHE.f FE ratio of CH4/C2H4 on Cux-CN catalysts at different potentials
To gain further insight into the improved CH4activity and selectivity and elucidate the role the atomic Cu sites in the Cu0.05-CN catalyst, temperature-programmed CO desorption and control experiments were carried out.The larger desorption peak area and stronger chemisorption of CO on the Cu0.05-CN catalyst than CN indicate the increased CO binding sites and enhanced CO binding strength resulted by Cu incorporation (Fig.S16a) [36].To probe the role of the single Cu atoms for CO2RR-to-CH4, the Cu0.05-CN catalyst was washed by 1.0 M nitric acid for 12 h at room temperature to obtain 0.05-CN sample and the almost all of the Cu metal was removed by acid treatment (Table S1).Compared with the Cu0.05-CN catalyst, the 0.05-CN catalyst exhibits much lower current density without CH4product at -1.2 VRHE(Fig.S16b-c).In addition, the effect of carbon black in the preparation of catalysts ink on the CO2RR is also excluded (Fig.S16d).Therefore, it is believed that the high CO2RR-to-CH4activity and selectivity on the Cu0.05-CN catalyst are originated from the single Cu sites embedded into the nitrogen cavities in the g-C3N4host.However, it should be noted that the semiconductor characteristic of g-C3N4host lead to the poor charge transfer ability of the Cux-CN catalysts, although the charge transfer resistance of CN can be reduced slightly by Cu doping (Fig.S17) [30].Therefore, improving the conductivity may be a promising for further boosting the CO2RR performance of Cux-CN catalysts.
3.3 Discussions
It has been reported that the isolated Cu sites tended to convert CO2into C1products in CO2RR, such as CO, CH4,and CH3OH [16, 19, 25, 59].On the other hand, the C2+products could also be obtained on Cu single-atom catalysts if the atomically dispersed Cu reversibly transformed into clusters or nanoparticles under reaction conditions or additional active centers acted collaboratively with single Cu sites [21, 22].Furthermore, the density of Cu sites and their distance played a key role in the selectivity between the CH4and the C2H4products and the ratio of CH4/C2H4as the adjacent Cu-N2sites enabled the C-C coupling,which is prerequisite for C2H4formation [25].Given two types of controllable and adjustable Cu sites in the Cux-CN catalysts in this work, the FE ratios of CH4/C2H4on Cux-CN catalysts are compared at different potentials.As shown in Fig.4f, the FE ratio of CH4/C2H4increased with the decrease of potentials for all catalysts.The Cu0.01-CN catalyst with the lowest Cu content and exclusive singleatom Cu sites shows the highest FE ratio of CH4/C2H4of 35.03 at -1.3 VRHE.Such high FE ratio of CH4/C2H4outperforms most reported results (Fig.S18), which is suggested to ascribed to the highly selective toward CH4of the single Cu atomic sites in nitrogen cavities and the long distance between these sites of the Cu0.01-CN catalyst with low Cu load.The Cu0.05-CN catalyst shows a CH4/C2H4FE ratio of 9.03 at -1.2 VRHE, at which the highest FE of CH4is achieved.The decreased CH4/C2H4FE ratio on the Cu0.05-CN catalyst is hypothesized to be resulted from the shortened distance between single Cu atoms at increasing Cu loads and the small amount of Cu2+species in the Cu0.05-CN catalyst [25].Therefore, the high CH4/C2H4FE ratio on the Cu0.01-CN catalyst and the outstanding CO2RR-to-CH4performance on the Cu0.05-CN catalyst support the hypothesis that the single Cu atoms embedded into the nitrogen cavities of the g-C3N4host are active sites for CH4formation.
To explore the reaction mechanism of the CO2RR on Cux-CN catalysts, in situ ATR-FTIR measurements were taken to identify the key intermediates during CO2RR in a homemade cell (Fig.S19).As shown in Fig.5a, the peaks at 1268 and 1350 cm-1can be ascribed to the *COOH intermediate, which is generally considered as the key intermediate for CO2electrochemical conversion to CO and further reduction [60].Notably, the signals at 1130 and 1490 cm-1, which can be assigned to *CH2O and *CH3O, respectively [61],intensify as the potentials decreased from -1.0 VRHEto -1.2 VRHE.Therefore, the formation of CH4on Cu0.2-CN catalysts is likely through the proton-electron transfer of *COOH,*CO, *CHO, *CH2O, and *CH3O in succession [60, 62].
To gain a comprehensive insight into the formation of atomic Cu sites and the structure-activity relationship of Cux-CN catalysts, DFT calculations were then performed.Previously, both experimental and theoretical studies have demonstrated that the nitrogen cavity consists of six nitrogen atoms in g-C3N4, which is the prevailing and stable structure for accommodating single metal atoms [36, 63].The calculation also shows that the formation energy of a Cu atom incorporated in one nitrogen cavity of g-C3N4(Cu1-g-C3N4) is -2.11 eV (Fig.5b),which is far less than that for forming two Cu atoms into one cavity (Cu2-g-C3N4).Furthermore, the experimental EXAFS spectrum can be fitted with great accuracy using Cu1-g-C3N4model, giving the average bond length R of 1.92 ? and the coordination number N of about 3.3 for Cu-N bond (Fig.S20 and Table S2).The optimized structure shows that the incorporated Cu atom coordinates with four N atoms in the nitrogen cavities and the charge transfer occurs from Cu to N atoms (Fig.S21).The calculated free energy profile demonstrates that the rate determining step (RDS) for the conversion of CO2-to-CH4is *CO → *CHO with a ΔG of 1.09 eV on the Cu1-g-C3N4single active site, which is about 0.14 eV lower than the ΔG of RDS of the C2H4pathway (Fig.5c).Therefore, it is more energy favorable to form CH4rather than other hydrocarbons on single Cu atomic sites hosted by g-C3N4.Based on the experimental observation, which shows the destructed g-C3N4structure and the increasing O-coordinated Cu atoms in the Cux-CN catalysts with increasing Cu loads, the model of Cu2-d-C3N4consisted of adjacent Cu sites and damaged C-N heterocycles is built (Fig.S22).The free energy barrier of the RDS for the CH4pathway is 0.11 eV higher than that for the C2H4pathway on the dual-site Cu2-d-C3N4(Fig.S23).According to the free energy differences between CH4and C2H4pathways(Fig.5d), it is concluded that the CH4pathway prevails over the C2H4pathway on single Cu atomic sites in nitrogen cavities of g-C3N4, while the trend reverses on dual Cu sites on destructed g-C3N4host.Therefore, the emergence of adjacent O-coordinated Cu sites in the Cux-CN catalysts with high Cu loads suppresses the CH4formation but facilitates the production of C2H4, as observed in experiments.Based on the mechanism and theoretical study, the CO2-to-CH4pathway on the favorable Cu sites is proposed to proceed via the intermediates of *COOH,*CO, *CHO, *CH2O, and *CH3O (Fig.5e).
4 Conclusions
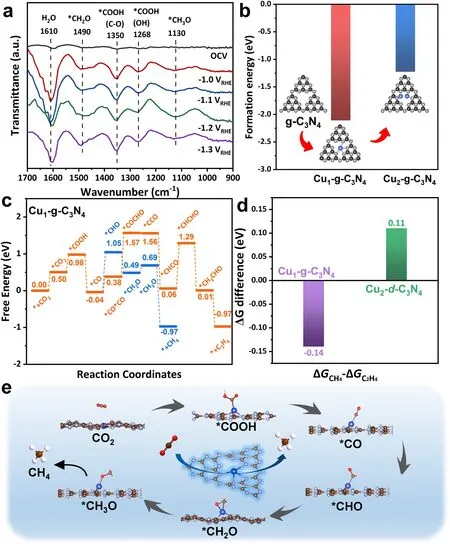
Fig.5 Mechanism and theoretical study.a In situ ATR-FTIR spectra of Cu0.05-CN catalysts at decreasing potentials in a CO2-saturated 0.1 M KHCO3 electrolyte.b Calculated formation energies of single and dual Cu sites in the nitrogen cavity of g-C3N4.c Free energy diagram of CO2 to CH4 and C2H4 on Cu1-g-C3N4.d Difference of free energy barriers between CH4 and C2H4 pathways on Cu1-g-C3N4 and Cu2-d-C3N4.e Proposed reaction pathway of CH4 formation on Cu1-g-C3N4
In this work, structurally stable single Cu atoms embedded in the nitrogen cavities of g-C3N4frameworks have been successfully synthesized with controlled configurations and chemical states to catalyze CO2RR for CH4production.High CH4activity and selectivity and high FE ratio of CH4/C2H4were achieved on the Cu0.01/Cu0.05-CN catalysts with predominant single Cu atomic sites.The increase of Cu contents in the Cux-CN catalysts leads to the emerging of a second O-coordinated Cu sites accompanied with the structural destruction of g-C3N4support, resulting in reduced CH4activity and selectivity and thus decreased CH4/C2H4ratio.This work provides a new strategy for constructing efficient single Cu atom catalysts for the conversion of CO2-to-CH4and delivers some insights into the understanding on the regulation of Cu-based catalysts for CO2RR to achieve desired selectivity and products.
AcknowledgementsThis work was supported by the Fundamental Research Funds for the Central Universities (2232021A-02 and 2232023Y-01) and the National Natural Science Foundation of China (Nos.52122312, 22209024 and 22202183).
Declarations
Conflict of interestThe authors declare no interest conflict.They have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Open AccessThis article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing,adaptation, distribution and reproduction in any medium or format,as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material.If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.To view a copy of this licence, visit http:// creat iveco mmons.org/ licen ses/ by/4.0/.
Electronic supplementary materialThe online version of this article (https:// doi.org/ 10.1007/ s40820- 023- 01188-1) contains supplementary material, which is available to authorized users.

- Nano-Micro Letters的其它文章
- Intelligent Vascularized 3D/4D/5D/6D-Printed Tissue Scaffolds
- Humanoid Intelligent Display Platform for Audiovisual Interaction and Sound Identification
- Initiating Binary Metal Oxides Microcubes Electromagnetic Wave Absorber Toward Ultrabroad Absorption Bandwidth Through Interfacial and Defects Modulation
- Effectively Modulating Oxygen Vacancies in Flower-Like δ-MnO2 Nanostructures for Large Capacity and High-Rate Zinc-Ion Storage
- Diverse Structural Design Strategies of MXene-Based Macrostructure for High-Performance Electromagnetic Interference Shielding
- Hetero Nucleus Growth Stabilizing Zinc Anode for High-Biosecurity Zinc-Ion Batteries