
分子動力學(xué)研究F1-ATP合酶對三磷酸腺苷的穩(wěn)定和定位作用
2015-12-05 06:30:14伍紹貴高曉彤徐成剛
物理化學(xué)學(xué)報(bào) 2015年9期
伍紹貴 高曉彤 李 權(quán) 廖 杰 徐成剛
(1四川師范大學(xué)化學(xué)與材料科學(xué)學(xué)院, 成都 610068;2中國科學(xué)院理論物理研究所理論物理國家重點(diǎn)實(shí)驗(yàn)室, 北京 100190)
分子動力學(xué)研究F1-ATP合酶對三磷酸腺苷的穩(wěn)定和定位作用
伍紹貴1,2,*高曉彤1李 權(quán)1廖 杰1徐成剛1
(1四川師范大學(xué)化學(xué)與材料科學(xué)學(xué)院, 成都 610068;2中國科學(xué)院理論物理研究所理論物理國家重點(diǎn)實(shí)驗(yàn)室, 北京 100190)
F1-ATP合酶通過與ATP之間建立廣泛的相互作用, 實(shí)現(xiàn)對ATP的位置進(jìn)行精確的定位. 這些相互作用為ATP的合成/水解創(chuàng)造了穩(wěn)定的環(huán)境. 理解這些相互作用是理解ATP的合成/水解機(jī)理的基礎(chǔ). 我們通過分子動力學(xué)模擬方法研究這些相互作用, 找出在穩(wěn)定化過程中起到重要作用的殘基. 通過檢測ATP和F1-ATP合酶之間的非鍵相互作用, 發(fā)現(xiàn)殘基段158–164所形成的loop區(qū)域及殘基R189, Y345對ATP存在顯著相互作用. 其中, 該loop區(qū)域?qū)TP的三磷酸部分形成一個(gè)半包圍結(jié)構(gòu), 封閉活性位點(diǎn)區(qū)域, 并通過氫鍵網(wǎng)絡(luò)約束ATP三磷酸的運(yùn)動, 為ATP合成/水解創(chuàng)造穩(wěn)定的環(huán)境. 此外, 關(guān)鍵殘基Y345通過π–π疊加相互作用對ATP的堿基進(jìn)行約束, 但是ATP的堿基可以在平行于Y345芳香環(huán)的平面內(nèi)進(jìn)行滑動, 我們推斷這種滑動運(yùn)動有利于促進(jìn)ATP的水解.
F1-ATP合酶; 氫鍵; 分子動力學(xué); 突變
1 Introduction
FOF1-ATP synthase is a central enzyme in the energy conversion that produces high-energy compound adenosine triphosphate (ATP) for all living organisms.1It synthesizes ATP from adenosine diphosphate (ADP) and inorganic phosphate (Pi) using the energy released by the passive flux of protons across the membrane down their electrochemical gradient.2Due to its biological importance, FOF1-ATP synthase receives extensive research interests both experimentally and theoretically.3FOF1-ATP synthase consists of two parts: FOand F1moieties. The water insoluble FOdomain is embedded in membrane and the hydrophilic F1domain protrudes into matrix space.4The F1moiety, usually termed as F1-ATPase, is a typical ATPase responsible for both ATP synthesis and hydrolysis. It is composed of five types of subunits (α, β, γ, δ, ε) in the stoichiometry (αβ)3γδε and only the β subunits are catalytically active. The β subunits have three different conformations: two closed (βDPand βTP) and one open (βE).5The βDPand βTPstates are similar in conformation while contain bound ADP and ATP, respectively. The βEis an empty conformation without nucleotide. ATP synthesis/hydrolysis reactions occur in a steady environment in the catalytic site to shield the interfluence of intense thermal oscillation. Thus these interactions between protein and ATP are essential to guarantee successful ATP synthesis/hydrolysis reaction. Understanding the interactions between ATP and F1-ATPase is essential for understanding ATP synthesis/hydrolysis.
Researchers have been focusing on how F1-ATPase works on a very efficient mode for more than 30 years.6To understand the basic interactions between substrate ATP and F1-ATPase is undoubtedly helpful for uncovering the ‘secret’ of the high efficient working mechanism of F1-ATPase. Alghouth experimental studies can solve high resolution structures of proteins with X-ray crystallography which provide a wealth of information, it is still chanllenging to obtain dynamics information from experiments at atomic resolution.7Protein dynamics are thought to be fundamental to protein function as protein structure.8Molecular dynamics (MD) simulations, a powerful complementary tool for experiments, can provide valuable dynamic information at atomic resolution.9By using physical force fields, all-atom MD simulations can reproduce detailed atomistic interactions between protein and ligands or between residues of protein.10These interactions can be analyzed by electrostatic interaction (Eele), van der Waals interaction (Evdw), hydrophobic contact, base-pairing, base stacking and so on. Therefore, computer simulations can complement experimental approaches by providing information inaccessible to experiments and helping address many remaining important questions.11
In the current study, high-resolution X-ray crystal structure of the catalytic β subunit in F1-ATPase adopting βTPconformation with bound ATP was used as the basic model for simulations. MD simulations were performed with F1-ATPase β subunit-ATP complex in explicit water to study the interactions between F1-ATPase and ATP. By analyzing the non-bonded interactions, we attempted to identify these critical residues which play important roles in positioning and stabilizing ATP in a catalytically competent conformation. It is expected that our study delivers a clear picture to describe F1-ATPase interacting with ATP at atomic scale.
2 Details of MD simulation
All-atom MD simulations were performed with the F1-ATPase β subunit-ATP complex in explicit water using GROMACS software, which is one of the fastest and most popular simulation packages available. Amber 03 force field12was used to describe protein residues and other Amber 03 force field parameters for ATP were developed by Meagher et al.13High resolution (0.19 nm) structure (PDB ID code 2JDI) reported by Walker et al.14was used as the basis for the F1-ATPase model. F1-ATPase consists of ~5400 residues and amounts to~26400 atoms in total. If the entire crystal structure is fully solvated in explicit water, the whole system will be computationally expensive for simulation. On the other hand, β subunit can serve as an ATPase to hydrolyze ATP independently. Therefore, only the β subunit with bound 5'-adenylyl-β-γ-imidodiphosphate (AMP-PNP) was separated from the original F1-ATPase crystal structure and the ligand AMP-PNP was modified to ATP by substituting the atom N connecting β and γ phosphates by an atom O. Additionally, the catalytic metal ion Mg2+and crystal water molecules were conserved for their important roles in forming essential interactions with protein or ligand (i.e. salt bridge/hydrogen bonds). The initial β subunit-ATP complex was solvated with 16199 three-point transferable intermolecular potential (TIP3P) water molecules15in a rectangular box of 6.73 nm × 7.82 nm × 10.94 nm. The distance from the surfaces of the box to the closest atoms of the solutes is ~1 nm, which is large enough to prevent F1-ATPase complex from contacting with its periodic images. All amino acid residues of F1-ATPase were set to protonation states at pH = 7. Sodium and chloride ions were added to make the system electrically neutral at a concentration of ~0.15 molL–1, close to the physiological ionic concentration. The final size of the system reaches~57000 atoms. A typical 1.0 nm cutoff was used for van der Waals and short-range electrostatic interactions. Long-range electrostatic interactions were treated using particle mesh Ewald (PME) method.16Berendsen barostat17and the velocity rescaling thermostat18were applied to control pressure and temperature at 1.01 × 105Pa and 300 K, respectively. A linear constraint solver (LINCS) algorithm was used to constrain all bonds involving hydrogen atoms.19Periodic boundary condition was applied in three dimensions of the simulation box. Motion equations were solved numerically with a time step of 2 fs and the neighbor list was updated every 10 steps. The solvatedsystem was firstly subjected to a thorough energy minimization using the steepest descent minimization method followed by a 200 ps MD simulation with position restrains on the heavy atoms of the solute. The final configurations were used for production simulations. To generate a representative ensemble of structures, each production simulation was performed for 100 ns and trajectories were saved at 2 ps intervals. The detailed conformation analysis and interaction calculations were conducted using the built-in tools of GROMACS. All the snapshots for protein-ligand complex were prepared with PyMOL (version 0.99) software.
3 Results and discussion
F1-ATPase makes extensive interactions with ATP through forming a network of hydrogen bonds/electrostatic interactions around ATP. Crystal structure shows that the bound ATP lies in a deep pocket of β subunit in F1-ATPase. The binding pocket of β subunit is responsible for the catalytically active reaction site. As shown in Fig.1, the binding pocket is enclosed by three helices (residues 156–178, residues 335–343, and residues 420–427) and two loops (residues 415–419 and residues 344–347). The triphosphate moiety of ATP is surrounded by a helix (residues 161–178) and a loop (residues 157–160). Owing to the charged nature of ATP triphosphate, multitude of hydrogen bond/electrostatic interactions are established between ATP triphosphate and F1-ATPase, which helps to seal off the catalytic site and leads to the closure of the previously open βEpocket.20
GROMACS has provided a set of trajectory analysis tools, which are convenient for molecular conformation characterization and energy measurement. Non-bonded interaction energy (Enon-bonded) can be determined by electrostatic interaction (Eele) and van der Waals interaction (Evdw), which is an easy, simple but effective approach to elucidate the interactions between two specified groups. In order to identify these residues which have important roles in positioning and stabilizing ATP, the nonbonded interactions between ATP and each residue of β subunit were determined. As shown in Fig.2A, residues 158–164, R189, Y345 have significant attractive interactions with ATP, with the strong to weak sequence of K162 > R189 > V164 > V160 > G161 > G159 > T163 > A158 > Y345 > F424. The loop segment (residues 158–164) surrounds ATP by a half and interacts with the β and γ phosphate groups through forming a network of hydrogen bonds to constraint the motion of ATP triphosphate. Residues G157, E188, E192, and D256 have a certain repulsive interactions with ATP, with the strong to weak sequence of E188 > G157 > D256 > E192. If the non-bonded interaction is decomposed into electrostatic interaction and van der Waals interaction, it is easy to find that the non-bonded interactions are mainly contributed by electrostatic interaction. In particular, Y345 interacts with ATP mainly through van der Waals interaction. As shown in Fig.1B, the spatial position distribution of these groups suggests that Y345 interacts with ATP base through π–π stacking interaction, which serves to constraint the position of ATP base. Additionally, due to the presence of the catalytic metal ion Mg2+, strong electrostatic interactions are existing between T163 and ATP triphosphate. Owing to the rich charged/polar natures of T163, the triphosphate group, and Mg2+, their interactions are very robust.
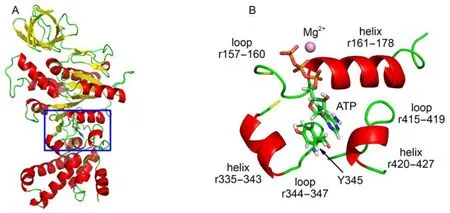
Fig.1 (A) X-ray crystal structure of F1-ATPase β subunit with bound ATP in βTPconformation, the box denotes the location of catalytic site; (B) enlarged drawing of the binding pocket involving the ATP and surrounding residues, Y345 interacting with ATP base through π–π stacking interaction
ATP is composed of three parts: a base (adenine), a ribose, and a triphosphate (Fig.3A). The numbers of hydrogen bonds formed between protein and each part of ATP were determined as shown in Fig.3B. The triphosphate moiety forms 7–12 hydrogen bonds with protein, which forms a hydrogen bond network between protein and the triphosphate moiety. These interactions seal off the catalytic site conformation and create a steady environment for ATP synthesis/hydrolysis. For other two parts, expect only one hydrogen bond between ATP base and protein occasionally, nearly no hydrogen bonds are formed. It suggests that the hydrogen bond interactions between F1-ATPase and ATP are chiefly located at the triphosphate moiety.
As above mentioned, due to the charged nature of ATP triphosphate-Mg2+complex, there exists multitude of hydrogen bond/electrostatic interactions between ATP's triphosphate and F1-ATPase, which form an interaction network to seal off the open conformation of the catalytic site. To obtain a clear picture of the interaction network, the hydrogen bond interactionsbetween the ATP triphosphate and the surrounding residues were examined as shown in Fig.S1, with a hydrogen bond existence map as given in Fig.S2. ATP's triphosphate moiety is surrounded by a helix (residues 161–178) and a loop (residues 157–160). The surrounding residues can be classified into two types according to the number of their hydrogen bonds formed with ATP triphosphate. The first type is single-hydrogen-bond residue, which forms only one hydrogen bond with the ATP triphosphate, such as G159, V160, G161, T163, and V164. The single hydrogen bond is formed between nitrogen atom in these residues and β or γ oxygen atom in ATP triphosphate, with normal occupancy values 60%–80%, such as N-HOB1on V160 (occupancy = 60.0% ± 6.8%), N-HOB1on G161 (occupancy = 73.7% ± 7.7%) and N-HOB2on T163 (occupancy = 77.7% ± 3.5%). The strongest hydrogen bond N-HOA2is formed on V164 with occupancy as high as 100% ± 0% while the weakest hydrogen bond N-HOG3is formed on G159 with occupancy = 37.1% ± 13.4%. The second type is multi-hydrogen-bond residue, such as K162, R189. Similar to single-hydrogen-bond residue, a robust hydrogen bond N-HOB1is formed between the nitrogen atom of K162 and β oxygen atom in ATP triphosphate with a high occupancy (97.1% ± 0.6%). The other hydrogen bonds on K162 are formed between the amino group of K162 and the ATP triphosphate. K162's amino group has three hydrogen atoms, which are able to alternatively form hydrogen bonds with β or γ oxygenatoms in ATP triphosphate due to the rotational feature of the bond Cε-Nζof K162. Another multi-hydrogen-bond residue is R189 with two amino groups, which can form robust hydrogen bonds with ATP triphosphate: Nη1-HOG2(occupancy = 62.9% ± 9.8%) and Nη2-HOG1(occupancy = 91.9% ± 6.0%). It should be noted that except hydrogen bond, hydroxyl oxygen OG1on T163 establishes strong electrostatic interactions with the atoms OB2, OG2on ATP triphosphate through a catalytic ion Mg2+.
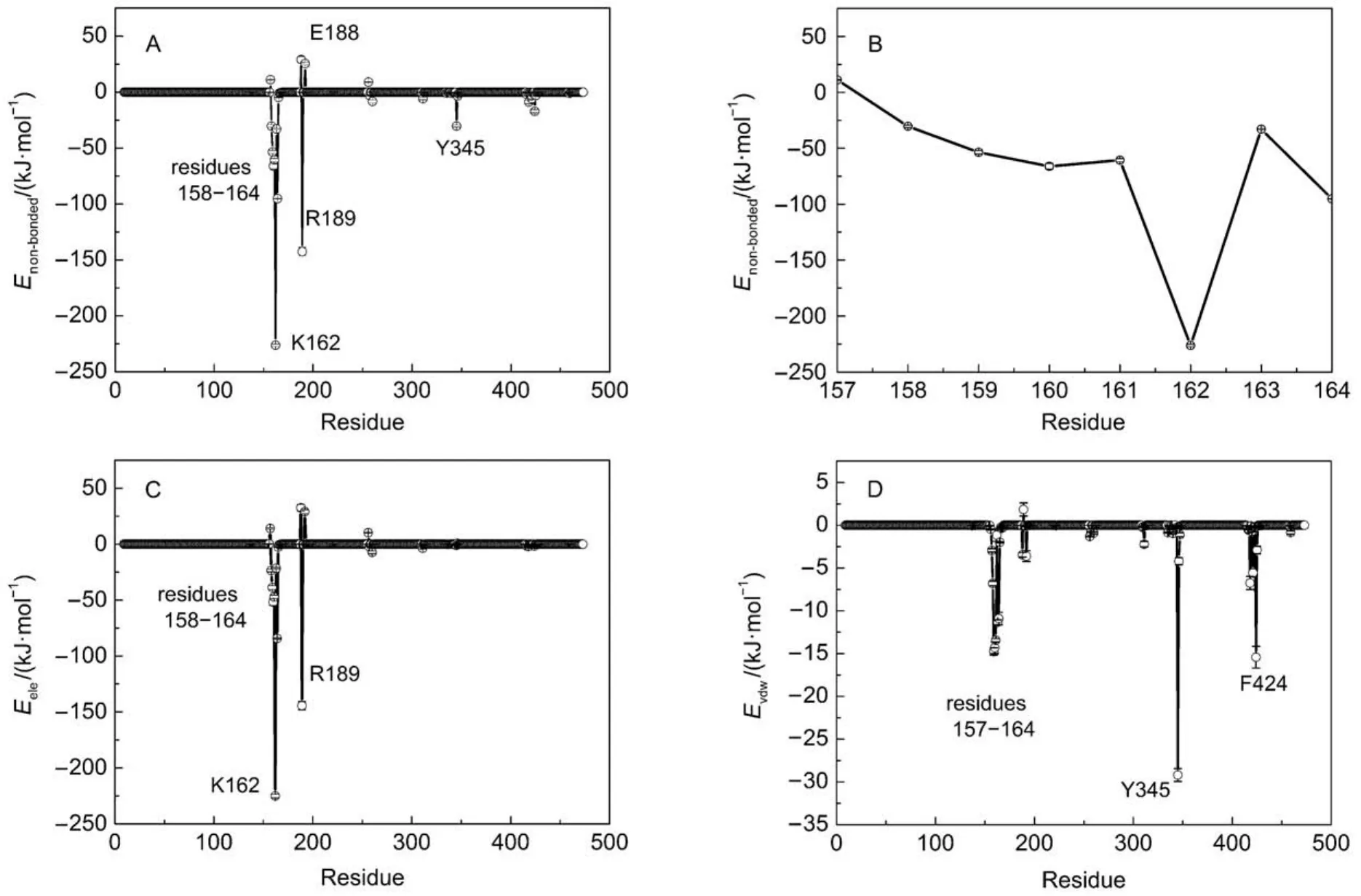
Fig.2 Interactions between F1-ATPase β subunit and ATP
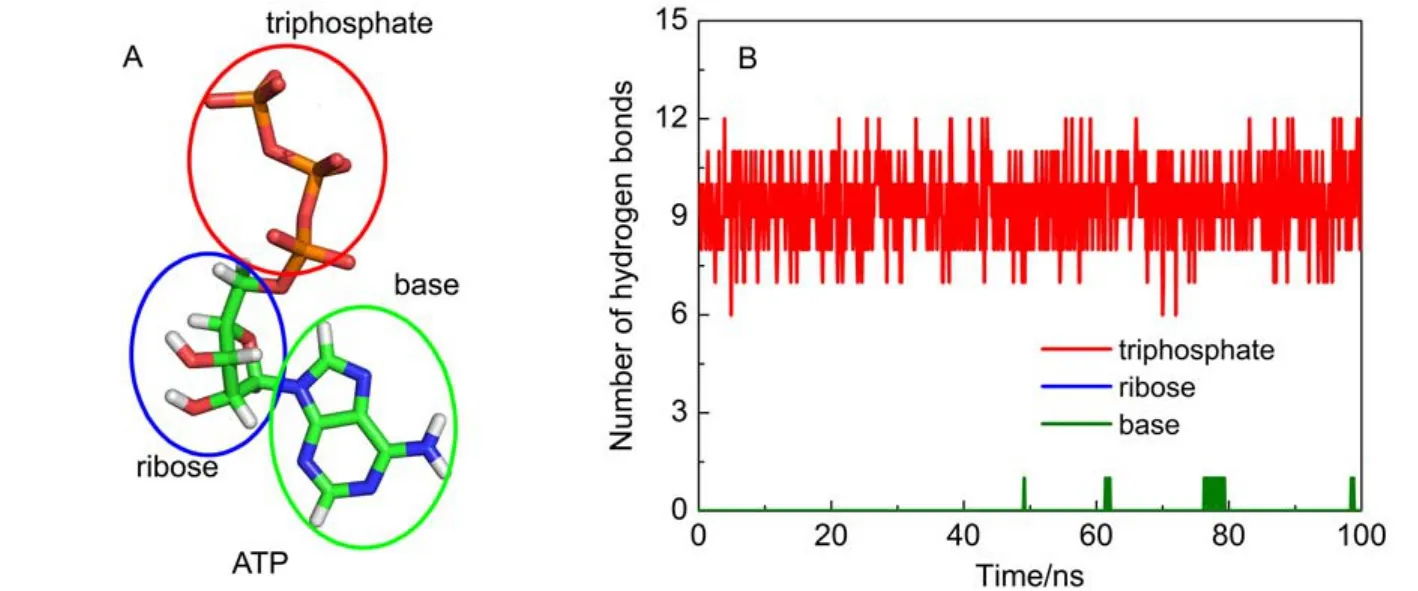
Fig.3 (A) ATP is composed of a triphosphate, a ribose and a base; (B) time evolution of hydrogen bond number for each component of ATP
As above mentioned, the ribose and base of ATP have nearly no hydrogen bond interactions with F1-ATPase. As shown in Fig.2D, a significant van der Waals interaction was detected between ATP base and Y345. As shown in Fig.4A, the spatial position distribution of these groups suggests that Y345 interacts with ATP base through π–π stacking interaction, which serves to position the ATP base. Single point mutations at Y345 were prepared as the starting configurations and MD simulations were carried out to further investigate the role of Y345. π–π stacking interactions are generally formed between aromatic groups. Considering the aromaticity of Y345's side chain, several aromatic amino acid residues were chosen to substitute for Y345, such as histidine (HIS), phenylalanine (PHE). HIS has a side chain of imidazole group which is aromatic under any pH value.21PHE is a nonpolar α-amino acid with an aromatic benzyl side chain. These mutated protein-ATP complexes were solvated and ionized following the same procedure as the WT system. MD simulations with each mutated system were carried out for 50 ns. For clarity, the distances between the center of masses (COMs) of ATP base and the aromatic groups of Y345, Y345F, and Y345H are referred as DY-base, DF-base, DH-baserespectively. Here, the time evolutions of these distances were monitored with their distributions as shown in Fig.4B. It is significant that DY-basefor WT system is small with a narrow distribution, suggesting the π–π stacking interaction between the hy-droxyphenyl group of Y345 and ATP base is robust. The DH-basedistribution curve for Y345H has one peak near 0.375 nm as well. However it has wider distribution with respect to the DY-basedistribution curve for WT system. This is attributed to the weak interaction between the imidazole ring of H345 and ATP base. A detailed examination of the MD simulation trajectory shows that the orientation of H345's imidazole ring can be easily deflected from the plane parallel to the ATP base, leading to a poor stacking on ATP base. Therefore the substitution of Y345 to HIS apparently decreases the stability of π–π stacking interaction in WT system. PHE has a similar stucture to TYR except one less hydroxy in the benzyl ring. Our simulations show the mutation Y345F can also establish π–π stacking interaction with ATP base. However, as shown in Fig.4B, the peak of DF-basecurve is located near 0.43 nm, a larger value than those of DY-base, DH-base, which means that a weaker π–π stacking interaction was established in the Y345F system. However, regardless of weak or strong interactions, these residues with aromatic side chains can establish π–π stacking interaction with ATP base. Compared with residue PHE, TYR has one more hydroxyl group. Although the additional OH group does not hydrogen bond with ATP, the above analysis suggests that it may help to strengthen the π–π stacking interaction between Y345 and ATP base.
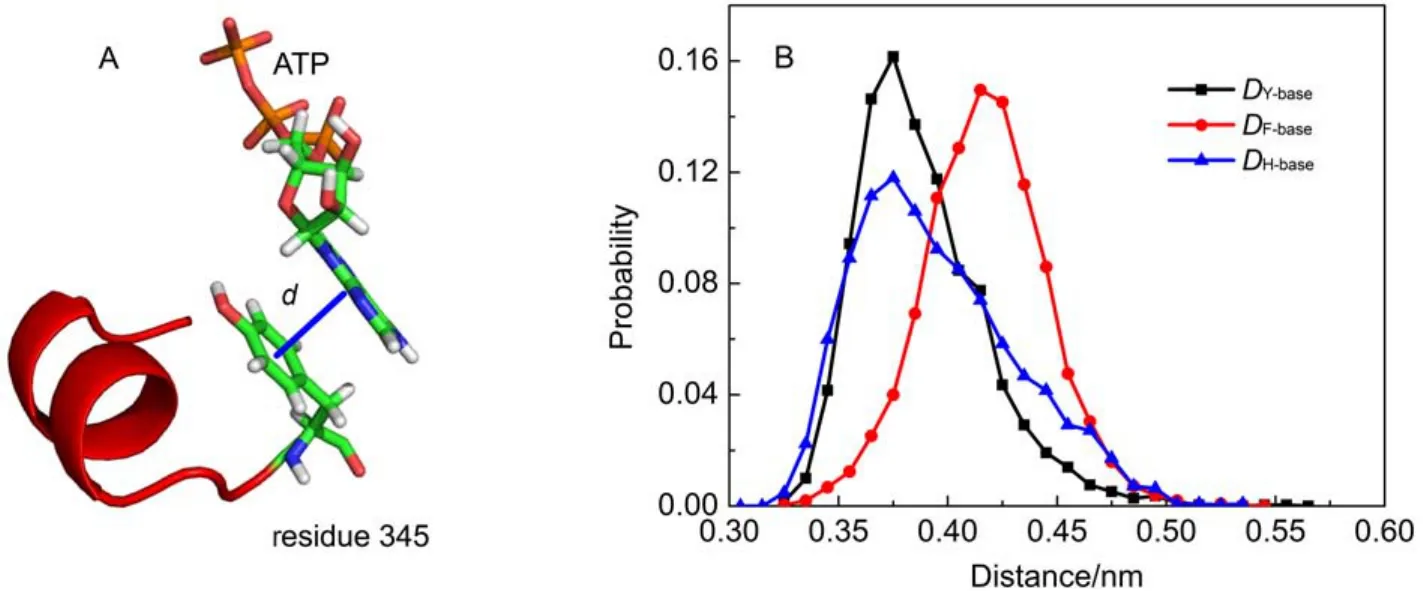
Fig.4 (A) Definition of distance between these π–π stacking interaction groups; (B) histograms of the distance distribution for DY-base, DF-base, DH-base
Additionally, the time evolution of the distance between the triphosphate and base of ATP, was determined as shown in Fig.5. It is interesting to find that at time t = 50, 60, 80 ns, many peaks are observed in the distance curve, suggesting that the distance between ATP triphosphate and its base is increased suddenly and then restores to its original value. The MD trajectory movie shows that these peaks are corresponding to the sliding motions of ATP base, which are still parallel to the aromatic ring of Y345. Known from the above analysis, protein almost does not impose constraint on ATP base through hydrogen bonds. Compared to electrostatic interaction, van der Waals interaction is a weak but essential interaction. Among these residues, Y345 has a strongest van der Waals interaction with ATP (Fig.2D). This interaction is mainly manifested as the π–π stacking interaction between Y345 aromatic ring and ATP base. It is evident that Y345 applies a two-dimensional constraint on ATP base and ATP base can make sliding motions parallel to Y345's aromatic ring. This motion can widen the distancebetween ATP triphosphate and base. On the other hand, the phosphate terminal of ATP is firmly held by the hydrogen bond network imposed by F1-ATPase. In view of the hydrolysis of ATP is the process to separate terminal phosphate from ATP, it is deduced that this motion may have a favorable contribution to ATP hydrolysis.
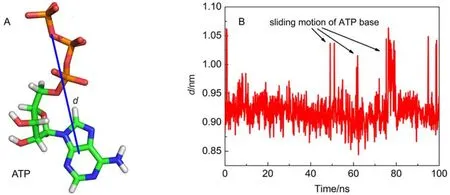
Fig.5 (A) Definition of the distance, d, between the COMs of ATP's triphosphate and base; (B) time evolution of d
4 Conclusion remarks
ATP synthesis/hydrolysis reactions occur in steady environments to shield the disturbance from thermal oscillation. F1-ATPase makes extensive interactions with ATP through forming a network of interactions around ATP. The interactions between protein and ATP provide a powerful guarantee for successful ATP synthesis/hydrolysis. Understanding these interactions between ATP and F1-ATPase is essential for understanding ATP synthesis/hydrolysis. In this work, all-atom MD simulations were performed using the βTPsubunit in F1-ATPase with bound ATP in explicit water to elucidate these interactions. By scanning the non-bonded interactions between ATP and each residue of the βTPsubunit in F1-ATPase, it is found that residues 158–164, R189, Y345 have significant interactions with ATP. The loop segment (residues 158–164) and R189 surround ATP by a half and they interact with the β and γ phosphates through forming a network of hydrogen bonds to constraint the motion of ATP triphosphate. The interaction network seals off the catalytic site conformation, and creates a steady environment for ATP synthesis/hydrolysis. In addition, ATP base is constrainted by the π–π stacking interaction from Y345. However, ATP base can make sliding motions parallel to Y345's aromatic ring. It is deduced that this motion may have relationship with ATP hydrolysis.
On the other hand, it is evidenced that ATP interacting with F1-ATPase adopts a different way from that in transcription. RNA polymerase adopts a three-component (hydrophobic contact, base stacking, and base pairing) mechanism for correctly positioning the incoming NTP base and the motion of the incoming NTP base is minimized. For ATP hydrolysis in F1-ATPase, ATP base is constrainted only by the π–π stacking interaction from Y345 and it can move in the two-dimensional plane parallel to Y345's aromatic group. The nucleotide moiety of NTP is incorporated into the 3' terminal of a nascent RNA with a pyrophosphate (PPi) which is released from the active site. For ATP hydrolysis in F1-ATPase, ATP is hydrolyzed into an ADP and a Piwhile both of them are released from the active site. Their resemblance lies in the fact that a steady environment is needed for chemical reactions. It is expected that our study throws light on the interactions between F1-ATPase and ATP and provides useful information for understanding the mechanism of ATP synthesis/hydrolysis.
Supporting Information: The snapshots of the hydrogen bonds between ATP triphosphate and F1-ATPase have been shown. Additionally, the existence map for these hydrogen bonds has been included. This information is available free of charge via the internet at http://www.whxb.pku.edu.cn.
(1)Ueno, H.; Suzuki, T.; Kinosita, K.; Yoshida, M. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 1333. doi: 10.1073/pnas.0407857102
(2)(a) Mitchell, P. Nature 1961, 191, 144. doi: 10.1038/191144a0 (b) Rastogi, V. K.; Girvin, M. E. Nature 1999, 402, 263.
(3)(a) Abrahams, J. P.; Leslie, A.; Lutter, R.; Walker, J. E. Nature 1994, 370, 621. doi: 10.1038/370621a0 (b) Zhou, Y.; Duncan, T. M.; Cross, R. L. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 10583. (c) Okuno, D.; Iino, R.; Noji, H. J. Biochem. 2011, 149, 655. (d) Iino, R.; Hasegawa, R.; Tabata, K. V.; Noji, H. J. Biol. Chem. 2009, 284, 17457.
(4)Al-Shawi, M. K.; Nakamoto, R. K. Biochemistry 1997, 36, 12954. doi: 10.1021/bi971477z
(5)Masaike, T.; Mitome, N.; Noji, H.; Muneyuki, E.; Yasuda, R.; Kinosita, K.; Yoshida, M. J. Exp. Biol. 2000, 203, 1.
(6)Dittrich, M.; Schulten, K. J. Bioenerg. Biomembr. 2005, 37, 441. doi: 10.1007/s10863-005-9487-7
(7)Da, L. T.; Avila, F. P.; Wang, D.; Huang, X. PLOS Comput. Biol.2013, 9, e1003020.
(8)(a) Moustafa, I. M.; Shen, H.; Morton, B.; Colina, C. M.; Cameron, C. E. J. Mol. Biol. 2011, 410, 159. doi: 10.1016/j.jmb.2011.04.078(b) Hammes-Schiffer, S.; Benkovic, S. J. Annu. Rev. Biochem. 2006, 75.
(9)Zhang, H.; Lu, J. R.; Mu, J. B.; Liu, J. B.; Yang, X. Y.; Wang, M. J.; Zhang, R. B. Acta Phys. -Chim. Sin. 2015, 31, 566. [張 賀,盧俊瑞, 穆江蓓, 劉金彪, 楊旭云, 王美君, 張瑞波. 物理化學(xué)學(xué)報(bào), 2015, 31, 566.] doi: 10.3866/PKU.WHXB 201501061
(10)Ai, Y. X.; Lu, J. R.; Xin, C. W.; Mu, J. B.; Yang, X. Y.; Zhang, H. Acta Phys. -Chim. Sin. 2015, 30, 559. [艾義新, 盧俊瑞, 辛春偉, 穆江蓓, 楊旭蕓, 張 賀. 物理化學(xué)學(xué)報(bào), 2014, 30, 559.]. doi: 10.3866/PKU.WHXB201401132
(11)Wu, S. G.; Sun, T.; Zhou, P.; Zhou, J. Acta Phys. -Chim. Sin. 2012, 28, 978. [伍紹貴, 孫 婷, 周 萍, 周 俊. 物理化學(xué)學(xué)報(bào), 2012, 28, 978.] doi: 10.3866/PKU.WHXB201202142
(12)Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M. C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T. J. Comput. Chem. 2003, 24, 1999.
(13)Meagher, K. L.; Redman, L. T.; Carlson, H. A. J. Comput. Chem. 2003, 24, 1016. doi: 10.1002/jcc.v24:9
(14)Bowler, M. W.; Montgomery, M. G.; Leslie, A. G.; Walker, J. E. J. Biol. Chem. 2007, 282, 14238. doi: 10.1074/jbc.M700203200
(15)Miyamoto, S.; Kollman, P. A. J. Comp. Chem. 1992, 13, 952.
(16)(a) Essmann, U.; Perera, L.; Berkowitz, M. L.; Darden, T.; Lee, H.; Pedersen, L. G. J. Chem. Phys. 1995, 103, 8577. doi: 10.1063/1.470117(b) Darden, T.; York, D.; Pedersen, L. J. Chem. Phys. 1993, 98, 10089.
(17)Berendsen, H. J.; Postma, J. P. M.; van Gunsteren, W. F.; DiNola, A.; Haak, J. J. Chem. Phys. 1984, 81, 3684. doi: 10.1063/1.448118
(18)Bussi, G.; Donadio, D.; Parrinello, M. J. Chem. Phys. 2007, 126, 014101. doi: 10.1063/1.2408420
(19)Hess, B.; Bekker, H.; Berendsen, H. J.; Fraaije, J. G. J. Comput. Chem. 1997, 18, 1463.
(20)Oster, G.; Wang, H. Biochim. Biophys. Acta 2000, 1458, 482. doi: 10.1016/S0005-2728(00)00096-7
(21)Mrozek, A.; Karolak-Wojciechowska, J.; Kie?-Kononowicz, K. J. Mol. Struct. 2003, 655, 397. doi: 10.1016/S0022-2860(03)00282-5
F1-ATPase Stabilizes and Positions Adenosine Triphosphate Revealed by Molecular Dynamics Simulations
WU Shao-Gui1,2,*GAO Xiao-Tong1LI Quan1LIAO Jie1XU Cheng-Gang1
(1College of Chemistry and Material Science, Sichuan Normal University, Chengdu 610068, P. R. China;2State Key Laboratory of Theoretical Physics, Institute of Theoretical Physics, Chinese Academy of Sciences, Beijing 100190, P. R. China)
F1-ATPase makes extensive interactions with ATP through forming a network of interactions around ATP. These interactions create a steady environment for ATP synthesis/hydrolysis. Thus understanding these interactions between ATP and F1-ATPase is essential for understanding ATP synthesis/hydrolysis mechanism. We performed all-atom molecular dynamics (MD) simulations to elucidate these interactions and attempted to identify key residues which play important roles in stabilizing and positioning ATP. By examining the non-bonded energies between ATP and residues of βTPsubunit in F1-ATPase, it is found that residues 158–164, R189, Y345 have significant interactions with ATP. The loop segment (residues 158–164) and R189 surround ATP by a half and they interact with β and γ phosphates through forming a network of hydrogen bonds to constraint the motion of ATP triphosphate. The interaction network seals off the conformation of the catalytic site, creating a steady environment for ATP synthesis/hydrolysis. Additionally, ATP base is positioned by the π–π stacking interaction from Y345. However, ATP base can slide and move paralleling to the aromatic group of Y345. It is deduced that this motion may facilitate ATP hydrolysis.
F1-ATPase; Hydrogen bond; Molecular dynamics; Mutation
O641; Q641.12
10.3866/PKU.WHXB201508062
Received: April 17, 2015; Revised: August 6, 2015; Published on Web: August 6, 2015.
*Corresponding author. Email: wsgchem@foxmail.com; Tel: +86-28-84760802.
The project was supported by the National Natural Science Foundation of China (11405113), Science and Technology Plan of Sichuan Province, China (2010JY0122), and Science Research Fund of Sichuan Normal University, China (10MSL02).
國家自然科學(xué)基金(11405113), 四川省科技廳項(xiàng)目(2010JY0122)和四川師范大學(xué)科學(xué)研究基金(10MSL02)資助
? Editorial office of Acta Physico-Chimica Sinica
猜你喜歡
云南化工(2021年9期)2021-12-21 07:44:16
云南化工(2021年6期)2021-12-21 07:31:42
鴨綠江·華夏詩歌(2021年10期)2021-05-12 01:39:00
中華建設(shè)(2019年2期)2019-08-01 05:57:46
天然產(chǎn)物研究與開發(fā)(2018年10期)2018-11-06 07:43:46
桃之夭夭B(2018年6期)2018-09-14 10:55:38
物理化學(xué)學(xué)報(bào)(2018年6期)2018-03-08 03:45:49
西湖(2017年4期)2017-04-07 07:46:56
現(xiàn)代檢驗(yàn)醫(yī)學(xué)雜志(2014年3期)2014-02-02 02:42:24
物理化學(xué)學(xué)報(bào)(2013年1期)2013-07-25 09:12:04
