
Formulation and evaluation of co-prodrug of furbiprofen and methocarbamol
2017-01-19 08:30:29
Department of Quality Assurance,Bharati Vidyapeeth College of Pharmacy,Near Chitranagari,Shivaji University,Kolhapur 416013,Maharashtra,India
Formulation and evaluation of co-prodrug of furbiprofen and methocarbamol
Neela Bhatia*,Kiran Katkar,Snehal Ashtekar
Department of Quality Assurance,Bharati Vidyapeeth College of Pharmacy,Near Chitranagari,Shivaji University,Kolhapur 416013,Maharashtra,India
A R T I C L EI N F O
Article history:
Received 21 April 2015
Received in revised form 7 October 2015
Accepted 15 October 2015
Available online 5 November 2015
NSAIDs
Flurbiprofen
Skeletal muscle relaxant
Methocarbamol,Ester prodrug
RP-HPLC
Ulcerogenicity
The current work envisages synthesis of an ester prodrug of furbiprofen whereby its carboxylic group was condensed with a skeletal muscle relaxant methocarbamol,with the aim of synergistic activity of two drugs,avoid furbiprofen mediated gastro-intestinal damage and minimize the ulceration tendency of furbiprofen.The synthesized prodrug was characterized and confrmed by physicochemical and spectroscopic studies.Solubility and partition coeffcient studies indicated an increased lipophilicity and thus better suitability for oral administration than the parent drugs and the protein binding studies revealed a low protein binding capacity of the mutual prodrug.Subsequently,in-vitrohydrolysis was studied in different pH,simulated gastric fuid,simulated intestinal fuid and plasma and quantitative evaluation was performed by high performance liquid chromatography.It was found that the prodrug remained unhydrolyzed in the stomach after absorption however,underwent rapid cleavage by the esterases in blood to give the parent drug.Furthermore,the mutual ester prodrug was evaluated for its anti-infammatory,analgesic,skeletal muscle relaxation,ulcerogenic and total acid content activity and was found to possess comparable activity with that of the parent drugs.Microscopic structures of the stomach tissues revealed signifcant reduction in gastric ulcer formation of mice gastric mucosa as compared to parent carboxylic acid drug.
?2016 The Authors.Production and hosting by Elsevier B.V.on behalf of Shenyang Pharmaceutical University.This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
1.Introduction
NSAIDs are a vast group of medications that are highly praised nowadays for their triple action:analgesic,antipyretic and antiinfammatory activities.The nonsteroidal anti-infammatory drugs are the most widely prescribed and used drugs for rheumatologic as well as nonrheumatologic conditions,which include acute and chronic pain[1],biliary,ureteric colic[2],dysmenorrhea[3],fever,and other applications[4].In other words, NSAIDs help to relieve pain,lower high body temperature or fever and reduce infammation.These are more prescribed thanopioids because of no narcotic effect and dependence risk. NSAIDs block the production of substances in the body called prostaglandins that play a role in pain,infammation,fever, and muscle cramps and aches.At low doses,NSAIDs work essentially as pain relievers while at higher doses they can actually reduce the body’s infammatory response to tissue damage as well as relieve pain.
Almost all NSAIDs on long-term treatment inhibit the enzyme cyclo-oxygenase(COX)and production of prostaglandins[5].Continuous intake of NSAIDs results in irritation of the gastric mucosa and enhances ulceration by blocking protective action of the prostaglandins on gastric mucosa,causing ulcer formation not only in the stomach but also in the lower part of the esophagus and also in the duodenum.In general, the properties of NSAIDs that contribute to ulcerogenesis can be divided into two categories:topical irritancy and the suppression of prostaglandin synthase activity.In addition,NSAIDs tend to damage the mucosa due to the presence of acid in the stomach and in the duodenum and in some cases due to the presence ofHelicobacter pylori(H.pylori)[6].The gastrointestinal side effects of NSAIDs are generally attributed to direct and/ or indirect mechanisms.The direct contact effect results usually from a local irritation produced by the acidic group of the NSAIDs and local inhibition of prostaglandin synthesis in the GI tract.The indirect mechanism is due to a generalized systematic action occurring after prior absorption.
Prodrugs,the pharmacologically inactive derivatives of active drugs,are designed to maximize the amount of active drug that reaches its site of action,through manipulation of the physicochemical,biopharmaceutical or pharmacokinetic properties of the drug.Prodrug synthesis is one of the approaches to modify chemical and physical parameters of parent drugs to overcome their limitations[7].At the same time,biotransformation of a prodrug to its parent compound at its target site of activity may be used to achieve rate-controlled and timecontrolled drug delivery of the actives[8].The prodrug approach has the ability to keep promising new drug candidates alive through development and to improve the safety and effcacy of existing drug products.It is a very fruitful area of research and its introduction in human therapy has given successful results in improving the clinical and therapeutic effectiveness of drugs with undesirable side-effects[9–11].
The aim of designing a mutual prodrug is to overcome limitations of a parent drug that would otherwise hinder its clinical use.Gastric mucosal injury produced by NSAIDs is generally aggravated by the local irritation caused by acidic group of NSAIDs[12,13].The temporary masking of this group may offer relief to the patient suffering from GI irritation;hence,prodrug approach is the most suitable technique for this purpose.The current work deals with a potent anti-infammatory drug associated with GI irritation,furbiprofen,which was selected as a model drug for carboxylic acid derivatization.Another drug, methocarbamol,is also clinically prescribed for painful spasms associated in GI disorders as skeletal muscle relaxant[14].The rationale of this work was to couple furbiprofen with methocarbamol to achieve reduced GI irritation and related side effects along with a synergistic effect of both actives.Generally,these two drugs are prescribed in combination;however, this combination often reveals the side effects associated with each other.Coupling of both compounds as a hybrid drug or through a spacer known as a mutual prodrug can result in a potent anti-infammatory compound with reduction of the main adverse local effects related to the activity of the NSAID and skeletal muscle relaxant.
2.Materials and methods
2.1.Materials
NSAID furbiprofen and skeletal muscle relaxant methocarbamol were kindly provided by Sun Pharma,Mumbai and Synthochem Pvt.Ltd.,Hyderabad.The other reagents and solvents used were of analytical/spectroscopic/HPLC grade as needed.The reactions were monitored byTLC on precoated silica G plates using iodine vapor as detecting agent.Melting points were recorded using capillary tube method.IR spectra were obtained by a Shimadzu IR spectrophotometer using KBr pellets.1H NMR spectra were recorded using DMSO on a Bruker-300 AVANCE instrument.Chemical shifts are expressed asδ(ppm).HPLC analysis,i.e.in-vitrohydrolysis studies,were carried out using a Jasco HPLC system using mobile phase acetonitrile:water(80:20) and detection wavelength 228 nm with fow rate 1 ml/min.The protocol for the animal experiments performed was approved by the IAEC(Institutional Animal Ethics Committee)as registered under the Committee for the Purpose of Control and Supervision of Experiments on Animals(Reg.No.988/C/06/ CPCSEA);approval number(BVCPK/CPSCEA/IAEC/01/08).Activities were carried out at the Department of Pharmacology,Bharati Vidyapeeth College of Pharmacy,Kolhapur.
2.2.Synthesis of prodrug
The reaction for synthesis of prodrugs involved two steps.The frst step consisted of acid chloride formation and the second step gives formation of mutual prodrug(Fig.1).Acid chloride formation is initiated by reacting furbiprofen with thionyl chloride.The acid chloride of furbiprofen was synthesized by reacting with excess thionyl chloride and refuxed for 30 min in RBF.After completion of the reaction,the mixture was cooled to room temperature and the excess of the thionyl chloride was removed from the reaction mixture.The product(acid halide derivative of furbiprofen)was collected and then washed with water.
In the second step,the acid chloride of furbiprofen was treated with the other drug methocarbamol to form the mutual prodrug.A three-necked fask with a sealed stirrer was ftted with a refux condenser and a dropping funnel.0.77 mol of methocarbamol,0.84 mol of pure dimethyl aniline and 100 ml of anhydrous ether was placed in the fask.The stirrer was set in motion and the mixture was heated to gentle refux on a water bath.0.79 mol of redistilled acid chloride of furbiprofen was run at such a rate that moderate refuxing continued after heating was removed.When two-third of the acid chloride was introduced,the dimethyl aniline hydrochloride commenced to crystallize and then the mixture was refuxed vigorously.Subsequently,it was cooled on an ice bath and after the refuxing ceased,the remainder of the acid chloride was added.The mixture was then heated on a water bath for 1 h.It was cooledto room temperature,100 ml of water was added and the stirring was continued until all the precipitated solid has dissolved. Diethyl ether was added to the cooled mixture.The ether layer was separated and ether was removed from it by evaporation. The product was collected and washed with hot water then recrystallized by ethyl acetate.The yield was found to be 66%[15].
The synthesized compounds were subjected toTLC to check their purity.A solvent system ethanol:water(70:30)was used and iodine vapor was used as detecting agent.Melting point of parent drugs and synthesized furbiprofen and methocarbamol (FLP-MET)prodrug was determined by capillary tube method. The structures of the synthesized compounds were established by their spectral(IR,NMR and mass spectral)analysis.
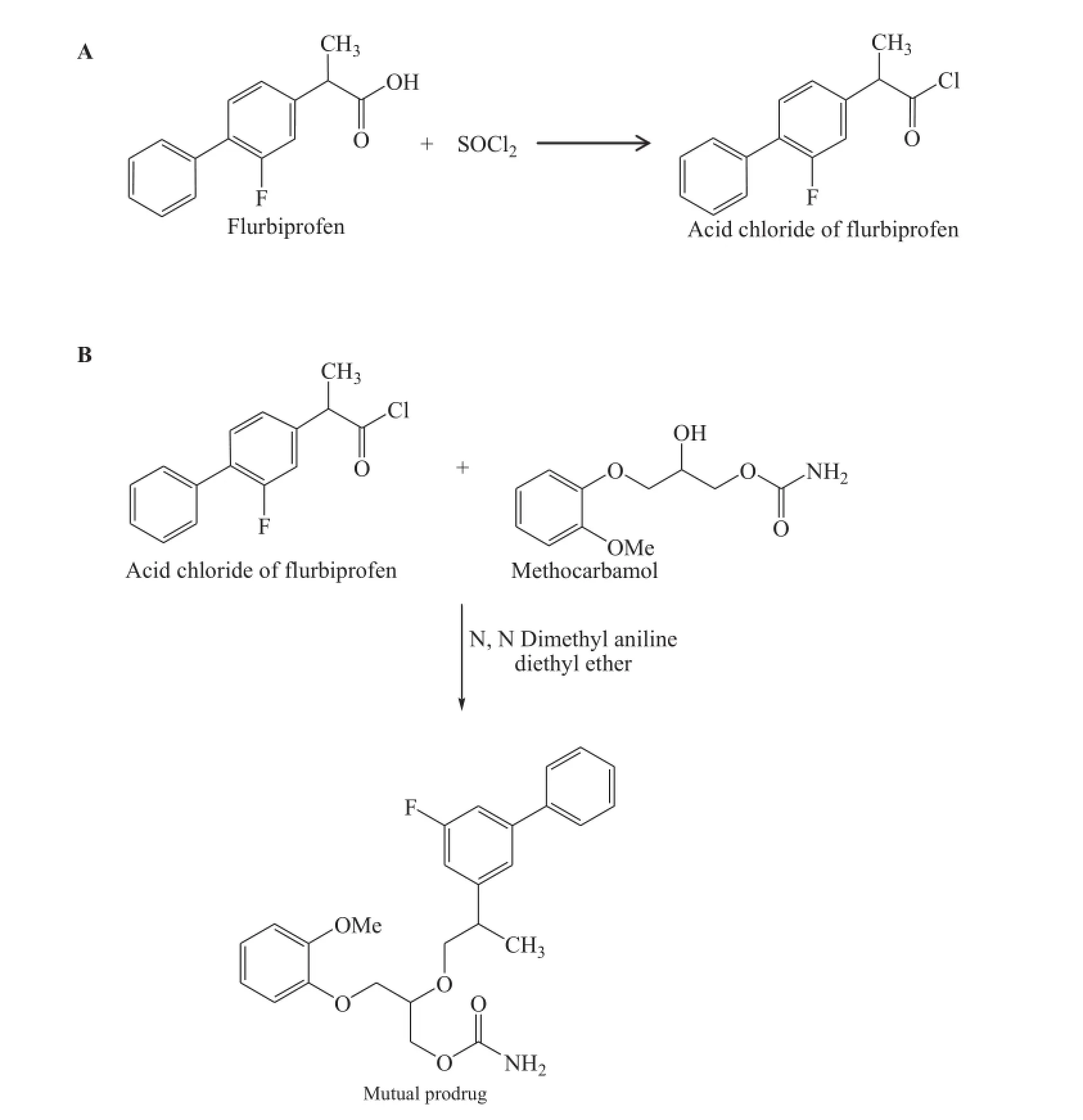
Fig.1–Scheme for synthesis of FLP-MET prodrug:(A)Acid chloride formation;(B)Synthesis of mutual prodrug from furbiprofen and methocarbamol.
2.3.Determination of solubility and partition coeffcient
Solubility of synthesized prodrug was determined in water, methanol,ethanol,chloroform,acetone and ether.Synthesized compounds were added to solvent(5 ml)in a vial that was tightly closed and subjected in a rotating shaker overnight at 60 rpm at room temperature,ensuring that equilibrium was established.The solutions were fltered through Whatman flter paper and fltrate was taken in an evaporating dish.The solvent was evaporated off and the weight of the residue was determined[16].
Partition co-effcient of the synthesized prodrug was determined in three systems viz.octanol:water,octanol:water(pH 1.2) and octanol:phosphate buffer(pH 7.4)at 25°C temperature.Synthesized compound(10 mg)was added to 10 ml of organic phase and 10 ml of aqueous phase was added to it.The mixture was shaken for 1 h and left for 2 h at 25°C.The two layers were separated out using a separating funnel.Prodrug concentration in aqueous phase,in hydrochloric acid buffer(pH 1.2)and phosphate buffer(pH 7.4),was determined from calibration curve plotted for the prodrug in the respective solvents[16,17].The partition coeffcient was calculated by Equation(1).

2.4.Protein binding studies
Stock solution of FLP,MET,FLP-MET prodrug each of concentration 10 mg/ml was prepared in phosphate buffer saline(PBS, pH 7.4).These solutions(100 ml)were taken in three separate beakers.Cellophane membrane with molecular weight cut off in the range 10,000–12,000 Dalton were frst washed with distilled water and then with buffer solution(pH 7.4)and tied at one end of the dialysis tube.Egg albumin(6%)was then flled into the dialysis tube through the other end and was dipped into the drug solutions and covered.The whole assembly was placed on a magnetic stirrer and switched at low rpm.The temperature was maintained at 37±0.5°C.After every hour,1 ml of PBS from three different beakers containing drug solutions were withdrawn and immediately replaced with fresh PBS. Withdrawn samples were diluted further with 1 ml PBS and concentration of those solutions was estimated using spectrophotometer at 248 nm for FLP,221 nm for MET and 247 nm for FLP-MET prodrug,respectively[18].
2.5.Hydrolysis study of mutual prodrug
Human plasma was procured from local blood bank to which 1 ml of acetonitrile was added and centrifuged for 15 min at 5000 rpm for separation of proteins.The supernatant was passed through the 45 mu syringe flter and used for hydrolysis study. The amount of drug that remained was determined by high performance liquid chromatographic analysis.HPLC analysis was done using HIQ Sil C-18 column(250 mm×4.6 mm,5 mu), using the mobile phase acetonitrile:water(80:20)and detection wavelength 228 nm with fow rate of 1 ml/min.
Stock solution of prodrug was prepared in methanol.Hydrolysis of mutual prodrug was studied under physiological conditions at different pH 3.0,4.0,5.0 and 7.4 in 0.05 M phosphate buffer at 37°C.Stock solution(10 ml)was added to 90 ml of preheated buffer solution in screw capped test tubes.To carry out hydrolysis study in plasma,2 ml of phosphate buffer(pH 7.4) was added to 8 ml of collected plasma at 37°C.The reaction was initiated by adding 1 ml of the stock solution of prodrug to 9 ml of preheated diluted plasma in a water bath at 37°C. To study the hydrolysis of synthesized prodrug in simulated gastric fuid(SGF,pH 1.2)and simulated intestinal fuid(SIF, pH 6.5)as per USP,the reaction was initiated by adding 1 ml of stock solution of prodrug to 9 ml of preheated SGF and SIF, which were maintained at 37°C.The reaction was done in a water bath at 37°C,and at appropriate intervals,1 ml sample was withdrawn and diluted with 9 ml of mobile phase and injected for chromatographic analysis[19,20].
After carrying out the hydrolysis study,the percent remaining concentration of the prodrug was calculated and then the rate constant(k)and half life(t1/2)values of the prodrug for different pH and plasma were calculated as in Equation(2).
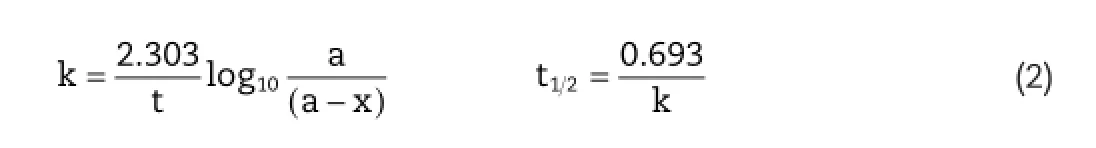
where k=specifc reaction rate constant;t=time for which hydrolysis is carried out;a=Initial concentration of prodrug; a?x=concentration of prodrug remaining at time t;t1/2=half life.
2.6.Anti-infammatory activity
The anti-infammatory activity of synthesized prodrug was evaluated using carrageenan-induced paw edema method on mice[11].Three albino mice with a body weight of 20–30 g were used for each test and control group.The animals were starved overnight.Before injection of the test compounds,the volume of the paw was measured plethysmographically by Vernier caliper.Animals were pretreated orally with drug solutions in the form of suspension prepared in 5%carboxy methyl cellulose.The control group received the same volume of the vehicle. Edema was induced after 30 min by sub-planter injection of 0.05 ml of a 1%solution of carrageenan solution in the animal’s left hind paw.The dose of furbiprofen(standard) 100 mg/kg and dose of FLP-MET prodrug(test)194 mg/kg was administered.The increase in paw volume was determined at 1,2,3,4,and 5 h after injection of the carrageenan;furbiprofen was used as standard for the test.The percent swelling inhibition was calculated using Equation(3).

whereVt andVo relate to the average volume in the hind paw of the mice before any treatment and after anti-infammatory agent treatment,respectively.Results were expressed as mean±SD.
2.7.Analgesic activity
Analgesic evaluation of the drugs was made using a previously reported method[21].Mice of either sex with weight 20–25 g were used.Acetic acid 0.01 ml in a concentration of 0.6% is injected intra-peritoneally.Groups of 3 animals were used for control and treated mice.The drug or the standard was administered intra-peritoneally to the test animals at various pretreatment times prior to acetic acid administration.The dose of furbiprofen(standard)100 mg/kg and dose of FLP-MET prodrug(test)194 mg/kg was administered.The mice were individually subjected into glass beakers and 5 min were allowed to elapse.The mice were then observed for a period of 10 min and the number of writhes was recorded for each animal.For scoring purpose,a writhe was indicated by stretching of the abdomen with simultaneous stretching of at least one hind limb.Percent inhibition was computed by Equation(4).
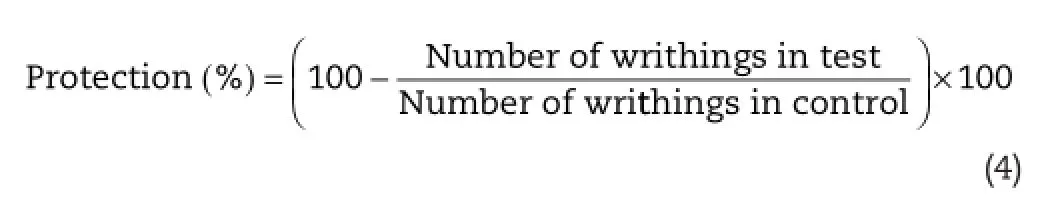
2.8.Skeletal muscle relaxant activity
The apparatus consisted of horizontal metal rod coated with rubber with 3 cm in diameter and attached to a motor with the speed adjusted to 2 rpm.The rod was 75 cm in length and was divided into 3 sections by plastic discs,thereby allowing the simultaneous testing of 3 mice.The rod was about 50 cm above the table top to discourage the animal from jumping off the roller.Mice with weight 20–30 g underwent a pretest onthe apparatus.Only those mice that demonstrated their ability to remain on the revolving rod for at least 1 min were selected for the test.The test compounds were administered for 30 min after intraperitoneal administration or 60 min after oral administration and the mice were placed for 1 min on the rotating rod.The dose of methocarbamol(standard)was 200 mg/ kg and dose of FLP-MET prodrug(test)was 388 mg/kg.The rota rod apparatus was adjusted at a speed of 25 rpm.The time taken by each animal to fall from the rotating rod was taken as a response(fall off time).An untreated mouse generally falls off within 3–5 min.Percent decrease in time was calculated using Equation(5)[22].
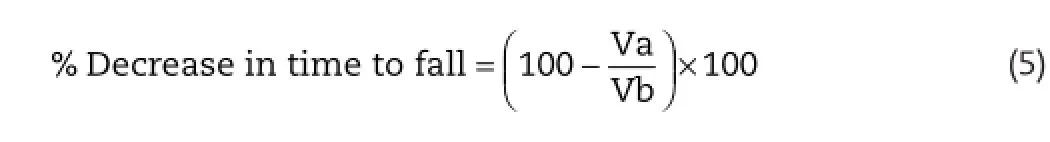
where Va=time taken by the animal to fall from the rotating rod after treatment with drug andVb=time taken by the animal to fall from the rotating rod before treatment with drug.
2.9.Ulcerogenic activity
Ulcerogenic activity was performed as per the protocol mentioned in literature[23].Groups of 3 Swiss albino mice with a weight 20–30 g were used for the control,standard and test compounds.The mice were fasted overnight with access to water. The doses were chosen considering the activity in the antiinfammatory tests in mice.The test compound(FLP-MET)was suspended in 0.1%methyl cellulose and given orally as 1 ml suspension to the frst group.Similarly,the second group was given equivalent dose of furbiprofen that serves as standard and the third group received equivalent amount of vehicle(CMC) that serves as control.The dose of furbiprofen(standard) 100 mg/kg and dose of FLP-MET prodrug(test)194 mg/kg was administered.The dosing was continued for two days.Food was withdrawn from all groups for 24 h after the last dose.The mice were then sacrifced 8 h post last dosing and their stomach were removed and placed on saline-soaked flter paper until inspection.A longitudinal incision along the greater curvature was made with fne scissors.The gastric contents were emptied for determination of total acid content.The stomach was inverted and the presence or absence of gastric irritation was determined.In the presence of single or multiple lesions(erosions,ulcers or perforation)was considered to be positive.The number of ulcers and hyperemia was noted.Ulcer index was calculated by Equation(6).

where x=total mucosal area/total ulcer area.
2.10.Total acid content
To determine the total acid content,the gastric contents were centrifuged at 1000 rpm for 10 min.0.1 ml of supernatant was diluted with 9 ml of distilled water and titrated against 0.01 N sodium hydroxide using 2 drops of phenolphthalein as indicator until permanent pink color was observed.The volume of NaOH required to give total acidity was noted.
Acidity(mEq/100 g)was calculated by Equation(7).
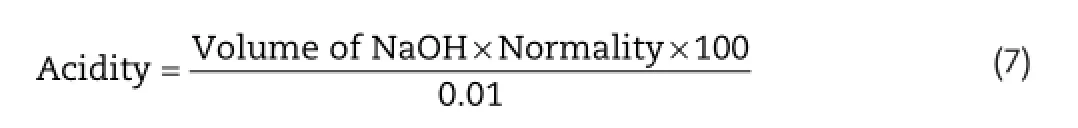
2.11.Statistical analysis
Statistical analysis of the pharmacological activity of the synthesized prodrugs on animals was evaluated using a oneway analysis of variance(ANOVA).Unpaired t-test was applied for expressing the signifcance and the experimental data are expressed as mean±SD.
3.Results and discussion
3.1.Physicochemical characterization
The product FLP-MET prodrug occurred as an offwhite-pale yellow powder with a yield of 59%.TLC profle of FLP,MET and FLP-MET in the eluent ethanol:water(70:30)gave Rf values of 0.82,0.92 and 0.88 respectively.The results of Rf value,melting point for synthesized FLP-MET prodrug were found to be different from parent drugs FLP,MET values indicating formation of new compound and suggesting completion of reaction.
FLP-MET prodrug:Melting point:114–117°C.Molecular weight:[M+]457.IR(KBr disk):γ(cm?1)3448.1,3327.57(N-H stretching),1681.62(C=O stretching),1259.29(C—O stretching).1H NMR(DMSO,300 MHz):δ 1.54–1.59(d,CH3);7.28(s,NH2); 6.912 to 7.58(m,aromatic hydrogens);3.7 to 4.32(m,methine and methylene protons);3.86(s,—OCH3).The ESI-MS shows the molecular ion peak at m/z 458.1(M+1).The results obtained from IR spectra,NMR and mass spectra(Fig.2) demonstrate the characteristic of the anticipated structure of the prodrug.
3.2.Solubility and partition coeffcient studies
The solubility of synthesized prodrug studied in different solvents is shown in Table 1.The FLP-MET prodrug was found to be more soluble in methanol and ethanol followed by acetone, chloroform,ether and very slightly soluble in water,thus indicating a non-polar nature of the synthesized prodrug.
For oral administration of a drug,the drug having octanol:water coeffcient–log P>2 is well absorbed,provided they have minimum solubility of 10 mg/ml.Results of lipophilicity study are shown in Table 2.The results indicated that the synthesized prodrug was relatively morelipophilic as compared to parent drugs,thus making the synthesized prodrug suitable for oral administration.Partition coeffcient results also suggested that mutual prodrug could cross the biological barrier to reach the systemic circulation.

Table 1–Solubility studies of FLP-MET prodrug.a
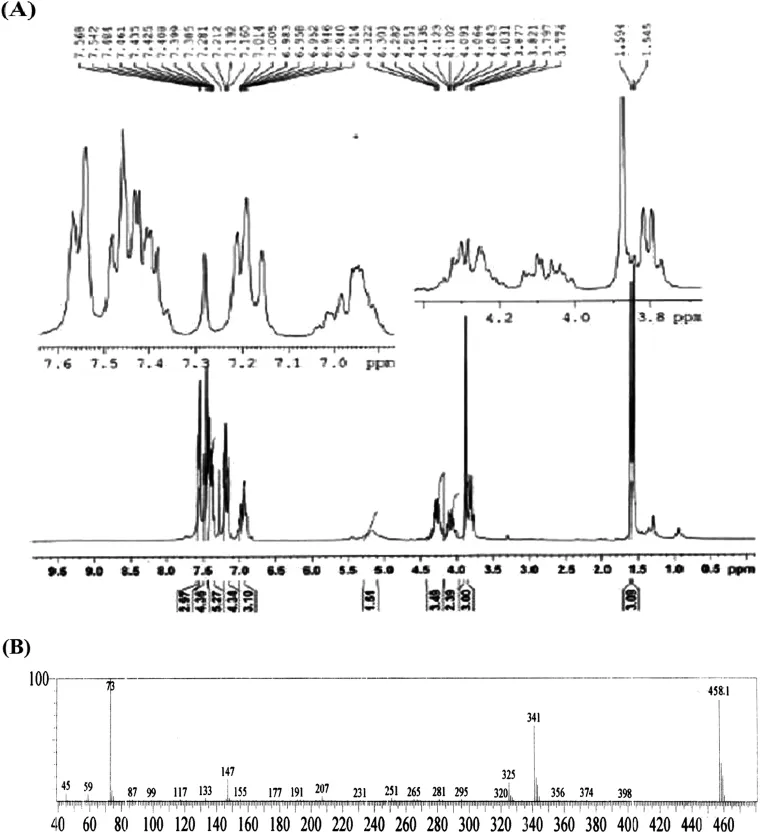
Fig.2–NMR and mass spectra of the FLP-MET prodrug.
3.3.Protein binding studies
Results obtained from protein binding studies indicated the low protein binding nature of the synthesized prodrug FLP-METas compared to the pure drugs furbiprofen and methocarbamol (Table 3).It was seen that the%concentration of unbound FLPMET prodrug was 44.82%as compared to 22.62%of unbound furbiprofen and 19.56%of unbound methocarbamol.As a result, more amount of free prodrug would be available at the target site and thus will be effective at low doses.

Table 2–Lipophilicity studies of FLP-MET prodrug.a
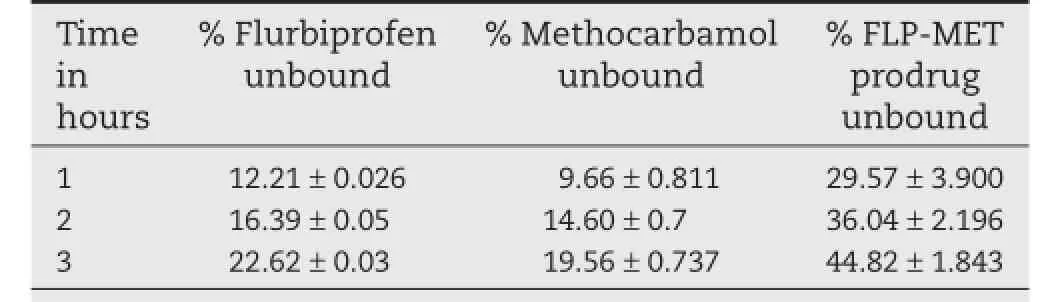
Table 3–Protein binding studies of FLP-MET prodrug at various time intervals.a
3.4.Hydrolysis study of mutual prodrug
Hydrolysis study was carried out to investigate the effect of certain factors like temperature,pH,SGF,SIF and plasma and enzymes on synthesized mutual prodrug.An HPLC method was developed to separate selectively the two drugs and the mutual prodrug.Fig.3 shows the HPLC chromatogram used for the quantifcation of un-hydrolyzed FLP-MET prodrug.Data related to the method development have not been presented.The kinetics of ester hydrolysis of the synthesized prodrug was studied in different buffer solutions having pH 3,4,5,7.4,and in SGF (pH 1.2)and SIF(pH 6.5).Also,the effect of activated esterase enzymes present in plasma was investigated to mimic the appropriate clinical range.Results presented in Table 4 indicated that a minimal amount of the prodrug hydrolyzed at gastric pH 3,4,5 and in SGF,SIF and also at the physiological pH 7.4. However the prodrug rapidly hydrolyzed in plasma,suggesting the activity of esterase enzyme present in the plasma acting on the prodrug.
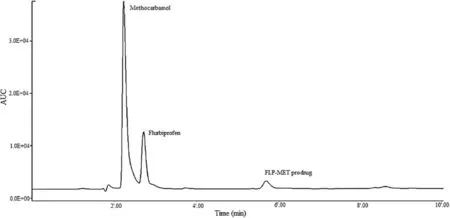
Fig.3–HPLC chromatogram indicating separation of FLP-MET prodrug from parent drugs.
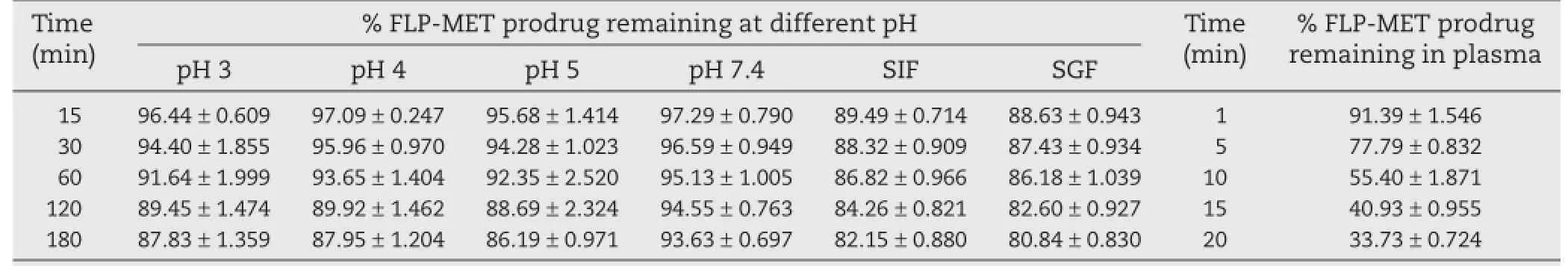
Table 4–Hydrolysis study of FLP-MET prodrug in different conditions.a
The reaction kinetics for degradation of the ester prodrug to its parent compounds was of frst order and a quantitative conversion to FLP and MET was observed by HPLC analysis. From Table 5,it is clear that the t1/2of the mutual prodrug of FLP-MET at various pH from 3 to 7.4 simulated gastric fuid and simulated intestinal fuid was found to be in the range of 8.4 to 13.54 h.However the t1/2of FLP-MET prodrug in plasma was found to be 4.42 min with a k-value of 1.566×10?1,whichstrikingly indicated that the drug underwent hydrolysis in presence of the esterases in the plasma.The data given in Table 5 followed a frst order kinetics for hydrolysis.The prodrug did not show signifcant hydrolysis at pH 3,4,5,7.4 and in SGF(pH 1.2)and SIF(pH 6.5),suggesting that the prodrug is relatively stable at this pH.The stability to pH hydrolysis implied that the compound can pass un-hydrolyzed through the stomach on oral administration and can be hydrolyzed by enzymes present in plasma releasing the parent compounds.

Table 5–k and t1/2values at different conditions for FLPMET prodrug.
3.5.Biological activities
The anti-infammatory activity was calculated as percentage inhibition of edema.The%inhibition of edema for mutual prodrug was comparable with parent NSAID(furbiprofen).Fig.4 demonstrates the results of anti-infammatory activity.A maximum anti-infammatory activity of mutual prodrug(FLPMET)was observed 83.75%at 4 h and 92.51%at 5 h.Statistical signifcance testing using unpaired t-test showed that the antiinfammatory activity of FLP-MET prodrug was comparable to that of furbiprofen(P<0.05).
From the comparative study between furbiprofen and the mutual prodrug,it is observed that the percentage of protection by FLP-MET prodrug against pain(writhing)induced by injection of irritant is comparable with the parent drug furbiprofen.The results given in Table 6 show the comparative analgesic activity of FLP and FLP-MET prodrug.
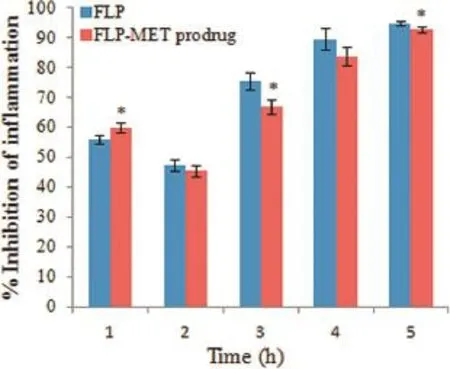
Fig.4–Comparison of anti-infammatory activity of FLP and FLP-MET prodrug.
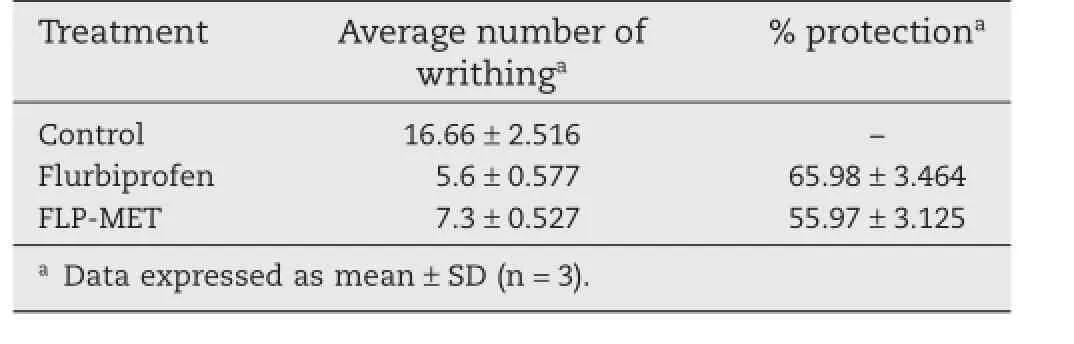
Table 6–Percent protection in acetic acid induced writhing by furbiprofen and FLP-MET prodrug.
Skeletal muscle relaxant activity was screened after intra-peritoneal dosing to the mice with control,test and standard drug.The results showed that skeletal muscle relaxant activity of prodrug was comparable to that of methocarbamol.Results of skeletal muscle relaxant activity are shown in Table 7.
Ulcerogenic activity of the synthesized prodrug was screened after treating the mice with equimolar quantity of furbiprofen and synthesized prodrug for 2 d.Ulcerogenic experiments in mice revealed that administration of furbiprofen over a period of 2 d resulted in a high incidence of gastric mucosa lesions shown by the ulcerative index which was found to be 26.9, whereas the results of the ulcerogenic activity of synthesized prodrug show that replacement of a carboxylic residue by an ester group could remarkably reduce the gastric ulceration(Table 7).At molecular equivalent quantity,the number of ulcers in gastric mucosa was markedly smaller in animals treated with the prodrug FLP-MET and ulcerative index was found to be 3.2.
The stomach specimen of the treated experimental animals under the microscope(biopsy)provided a precise method for investigation of the ulcerogenic potential of the NSAID and synthesized mutual prodrug.Fig.5 shows microscopic structures of the stomach tissues under the same magnifcation.As seen from the fgure,the group treated with the furbiprofen showed complete damage of mucous layer besides submucosal ulceration.These effects were not observed with the group treated with control.In the case of the prodrug,these effects are minimal.This observation affords good evidence for the safety of the suggested oral delivery system of prodrug compared withthe traditional use of the parent drug.Table 6 shows a comparison of ulcer index after administering test compound and standard drug.
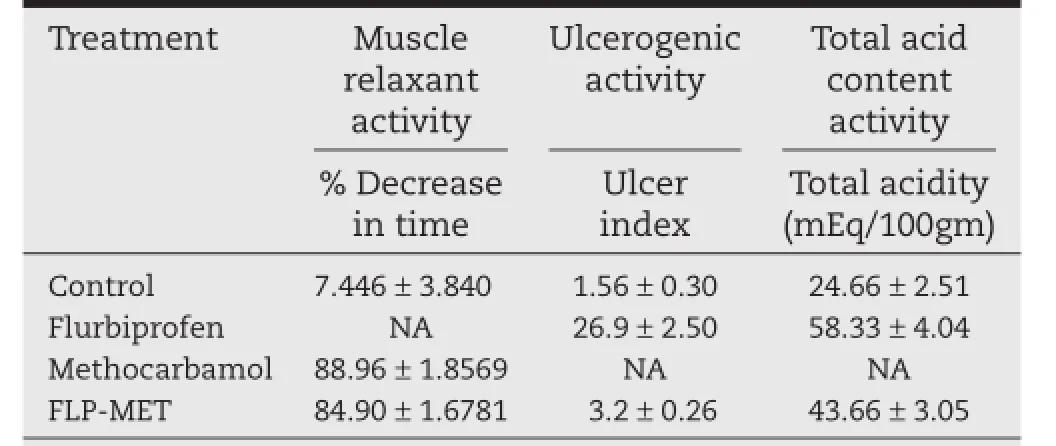
Table 7–Muscle relaxant activity,ulcerogenic activity and total acid content activity of furbiprofen, methocarbamol and FLP-MET prodrug in mice.a
The values of total acid content in the case of the mice treated with furbiprofen and the synthesized prodrug were determined.Results of total acid content activity are given in Table 7.The amount of acid secreted was found to be less in the case of the synthesized prodrug as compared to their parent NSAID furbiprofen,which indicates that the synthesized compound possesses less tendency to produce gastric acid as compared to standard drug furbiprofen,hence minimizing GI side effects.
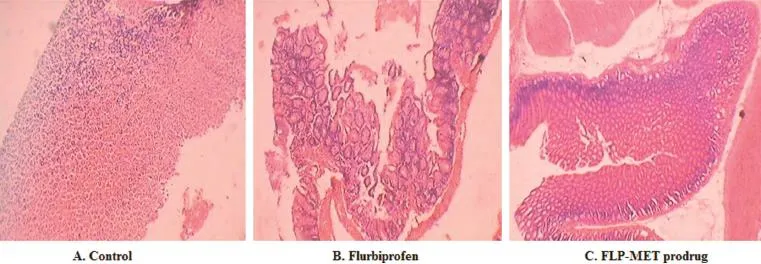
Fig.5–Effect of FLP and FLP-MET on microscopic sections of stomach.
4.Conclusion
A mutual prodrug was synthesized from the parent drugs furbiprofen and methocarbamol.Absorption bands obtained in IR and NMR spectrum confrmed the formation of ester linkage between furbiprofen and methocarbamol.The chemical and enzymatic hydrolysis studies of ester prodrug at different pH and in simulated gastric fuid and in simulated intestinal fuid did not show signifcant hydrolysis,suggesting that that the prodrug was quite stable at these pH values. However,the hydrolysis of prodrug in plasma indicates rapid hydrolysis,suggesting the activity of esterase enzyme present in the stomach acting on the prodrug showing the fulfllment of the prodrug design.Protein binding studies indicated the low protein binding nature of the synthesized prodrug as compared to the parent drugs,which translates to an increase in bioavailability of the prodrug and thus requirement of lower doses.In case of biological activities anti-infammatory, analgesic and skeletal muscle relaxant activity,the activity of the synthesized prodrug was found to be comparable with standard drugs.Ulcerogenic activity and total acid content activity studies showed that the ulcerogenic lesions and gastric acid produced by prodrug were less when compared to the parent drug furbiprofen,thus indicating that the prodrug formed has less ulceration potential and gastrointestinal side effects as compared to the parent drugs.
Acknowledgements
The authors express their thanks to Sun Pharma,Mumbai and Synthochem Labs Pvt.Ltd.,Hyderabad for kindly providing samples of furbiprofen and methocarbamol.
R E F E R E N C E S
[1]Portenoy R,Rosenblum A,Marsch L,et al.Opioids and the treatment of chronic pain:controversies,current status,and future directions.Exp Clin Psychopharmacol 2008;16:405–416.
[2]Sandle R,Christopher M,Alexandra C,et al.Nonsteroidal anti-infammatory drugs,apoptosis,and colorectal adenomas.Gastroenterology 2002;123:1770–1777.
[3]Dawood M.Nonsteroidal anti-infammatory drugs and changing attitudes toward dysmenorrhea.Am J Med 1988;84:23–29.
[4]Brooks P.Use and benefts of nonsteroidal antiinfammatory drugs.Am J Med 1998;104:9S–13S.
[5]Vane J,Botting R.Mechanism of action of anti-infammatory drugs.Scand J Rheumatol 1996;25:9–21.
[6]Wallace J.How do NSAIDs cause ulcer disease?J Clin Gastroenterol 2000;14:147–159.
[7]Jarkko R,Hanna K,Tycho H,et al.Prodrugs:design and clinical applications.Nat Rev Drug Discov 2008;7:255–270.
[8]Stella V,Charman W,Naringrekar V.Prodrugs.Do they have advantages in clinical practice?Drugs 1985;29:455–473.
[9]Clercq E,Field H.Antiviral prodrugs–the development of successful prodrug strategies for antiviral chemotherapy.Br J Pharmacol 2006;147:1–11.
[10]Doh H,Cho W,Yong C,et al.Synthesis and evaluation of Ketorolac ester prodrugs for transdermal delivery.J Pharm Sci 2003;92:1008–1017.
[11]Khan M,Akhter M.Synthesis,pharmacological activity and hydrolytic behavior of glyceride prodrugs of ibuprofen.Eur J Med Chem 2005;40:371–376.
[12]Makhija D,Somani R,Chavan A.Synthesis and pharmacological evaluation of antiinfammatory mutual amide prodrugs.Indian J Pharm Sci 2013;75:353–357.
[13]Dhikav V,Singh S,Pande S,et al.Non-steroidal druginduced gastrointestinal toxicity:mechanisms and management.J Indian Acad Clin Med 2003;4:315–322.
[14]Chou R,Peterson K,Helfand M.Comparative effcacy and safety of skeletal muscle relaxants for spasticity and musculoskeletal conditions:a systematic review.J Pain Symptom Manage 2004;28:140–175.
[15]Furniss B,Hannaford A,Smith P,et al.Vogel’s textbook of practical organic chemistry.New York:ELBS Publications; 1988.p.704.
[16]Mishra A,Veerasamy R,Jain P,et al.Synthesis, characterization and pharmacological evaluation of amide prodrugs of Flurbiprofen.J Braz Chem Soc 2008;19:89–100.
[17]More H,Hajare A.Practical physical pharmacy.India:Career Publications;2007.p.64–70,164-185.
[18]Rasheed A,Kumar C.Synthesis,hydrolysis and pharmacodynamic profles of novel prodrugs of mefenamic acid.Int J Curr Pharm Res 2009;1:47–55.
[19]Zhao X,Tao X,Wei D,et al.Pharmacological activity and hydrolysis behaviour of novel ibuprofen glucopyranoside conjugates.Eur J Med Chem 2006;41:1352–1358.
[20]Sharma P,Menon B.Design synthesis and evaluation of diclofenac-antioxidant mutual prodrugs as safer NSAIDs. Indian J Chem 2009;48:1279–1287.
[21]Dhaneshwar S,Dev S,Mhaske D,et al.Synthesis and pharmacological evaluation of cyclodextrin conjugate prodrug of mefenamic acid.Ind J Pharm Sci 2007;69:69–72.
[22]Vogel G.Drug discovery and evaluation,pharmacological assay.Germany:Springer;2002.p.398,759,770.
[23]Sheha M,Khedra A,Elsheriefb H.Biological and metabolic study of naproxen–propyphenazone mutual prodrug.Eur J Pharm Sci 2002;17:121–130.
*< class="emphasis_italic">Corresponding author.
.Department of Quality Assurance,Bharati Vidyapeeth College of Pharmacy,Near Chitranagari,Shivaji University,Kolhapur 416013,Maharashtra,India.Tel.:+91 231 2637286;fax:+91 231 2638833.
E-mail address:neela.bhatia20@gmail.com(N.Bhatia).
Peer review under responsibility of Shenyang Pharmaceutical University.
http://dx.doi.org/10.1016/j.ajps.2015.10.031
1818-0876/?2016 The Authors.Production and hosting by Elsevier B.V.on behalf of Shenyang Pharmaceutical University.This is an open access article under the CC BY-NC-ND license(http://creativecommons.org/licenses/by-nc-nd/4.0/).
 Asian Journal of Pharmacentical Sciences2016年3期
Asian Journal of Pharmacentical Sciences2016年3期
- Asian Journal of Pharmacentical Sciences的其它文章
- A novel LC–MS/MS assay for methylprednisolone in human plasma and its pharmacokinetic application
- Auricularia auricular polysaccharide-low molecular weight chitosan polyelectrolyte complex nanoparticles:Preparation and characterization
- Brain targeted delivery of paclitaxel using endogenous ligand
- Generic sustained release tablets of trimetazidine hydrochloride:Preparation and in vitro – in vivo correlation studies
- Effect of process and formulation variables on the preparation of parenteral paclitaxel-loaded biodegradable polymeric nanoparticles: A co-surfactant study
- Preparation and evaluation of PEGylated phospholipid membrane coated layered double hydroxide nanoparticles