
Adult-onset mitochondrial encephalopathy in association with the MT-ND3 T10158C mutation exhibits unique characteristics: A case report
2019-08-14 01:37:10XiaoLiFuXiangXueZhouZhuShiWeiChengZheng
World Journal of Clinical Cases 2019年9期
Xiao-Li Fu, Xiang-Xue Zhou, Zhu Shi, Wei-Cheng Zheng
Abstract
Key words: Mitochondrial disease; Stroke-like episode; Magnetic resonance; diagnosis;Case report
INTRODUCTION
Diagnosis of mitochondrial disease is challenging. Symptoms such as a stroke-like episode, lactic acidosis, deafness, diabetes mellitus, short stature, and myopathy, often with a family history showing maternal inheritance, are the most common characteristics of a mitochondrial disease. Additionally, a stroke-like episode often occurs in childhood or adolescence. Nonetheless, a diagnosis of mitochondrial diseases in patients with adult onset is often delayed due to a lack of family history, atypical manifestations, and limited resources for functional, pathologic, and/or genetic testing in routine clinical practice[1]. We report an adult-onset patient with mitochondrial encephalopathy carrying the m.10158T >C mutation in theMT-ND3gene. The encephalopathy manifested as isolated recurrent stroke-like episodes without clinical myopathy or other organ dysfunction. In this paper, we focus on how we diagnosed this case and the distinct feature of the brain magnetic resonance imaging (MRI), which is important for preventing misdiagnosis and delay in treatment. Additionally, we summarize the clinical implications of such cases with this mutation.
CASE PRESENTATION
Chief complaints
A 52-year-old female presented with a sudden onset of right-sided numbness and weakness that was accompanied by a left temporal cluster-like headache. No fever or prodromal infection was found at disease onset.
Personal and family history
The family and personal history was unremarkable.
Physical examination upon admission
On physical examination, the height and weight of the patient were 154 cm and 56 kg,respectively. Vital signs were normal, as were heart, lung and abdominal examinations. Neurological examination showed intact mental status, with normal speech and comprehension. Mild 4/5 right-sided hemiparesis was present with normal tone in both the arm and leg, though no other focal neurological deficits were found. After admission, she complained of discomfort and tingling in the right leg, after which a generalized tonic-clonic seizure for 3 min occurred before it was stopped by a bolus of intravenous diazepam.
Laboratory examinations
Laboratory tests, including D-dimer, lactic acid, and serum autoantibody levels, as well as thyroid function and tumor markers indicated no apparent abnormalities.Glucose tolerance and lactic acid movement tolerance tests were normal. A lumbar puncture was performed, and her open intracranial pressure was 180 mm H2O.Cerebrospinal fluid (CSF) testing showed that cell counts and protein, glucose,chloride, monoclonal antibody, adenosine deaminase, and lactate dehydrogenas levels were within normal ranges.
Imaging examinations and history of present illness
MRI demonstrated a lamellar left parietal lobe lesion predominantly involving the cortex, with hyperintensity on both diffusion-weighted imaging and fluid-attenuated inversion recovery (Figure 1). The apparent diffusion coefficient map revealed a preserved, isointense signal. No abnormalities were found by susceptibility weighted imaging or magnetic resonance angiography and venography (Figure 1). Due to the stroke-like onset pattern and MRI features, further thrombophilia screening was performed and showed decreased protein S activity. A diagnosis of cortical venous thrombosis was first proposed. Mitochondrial encephalomyopathy, lactic acidosis,and stroke-like episodes (MELAS) was also considered but temporarily excluded because of the incomplete manifestation and lack of genetic evidence. Anticoagulation therapy was initiated, and follow-up was performed to maintain the international normalized ratio (INR) within the target range.
Two months later, the patient was readmitted for subacute cognitive impairment.She was unable to identify and communicate with family members; she also had difficulty understanding questions or instructions and instead responded by repeating the word "nothing". During hospitalization, a secondary generalized seizure occurred, initially with eyes gazing to the right and then convulsion developing,which lasted for approximately 10 s before self-alleviation. Neurological examination suggested transcortical sensory aphasia, with fully covered limb strength. Blood tests and CSF examination were normal; INR was 2.21. On repeated MRI, new lesions were identified in the left temporal lobe and were also detected 10 days later in the right temporal lobe on radiological follow-up (Figures 1 and 2). Although the MRI signal characteristics are consistent with the initial findings, the original lesion in the left parietal lobe had been alleviated, with cortical atrophy. We further conducted magnetic resonance spectroscopy (MRS), which revealed markedly elevated lactate(Lac) concentrations in the regions of interest in the left temporal lesion (Figure 1).Mitochondrial encephalopathy was diagnosed, and genetic testing using peripheral blood was performed. However, DNA testing for frequent MELAS and myoclonic epilepsy with ragged red fibers syndrome mutations were negative. Because of the lack of symptoms of muscle weakness or pain, the patient declined our suggestion of performing a muscle biopsy. Anticoagulation therapy was terminated, and levetiracetam (1000 mg/d) was administered.
At 3 mo after her second admission, the patient was experiencing involuntary movement in her left limbs, with repetitive flexion/extension. An MRI scan showed a hyperintense signal abnormality in the right parietal lobe (Figure 2). Brachial biceps biopsy was performed. Histopathology revealed no abnormalities, and no necrotic or regenerating fibers were observed; ragged-red fibers and intense succinate dehydrogenase activity were not detected. Nonetheless, complete sequencing of mitochondrial DNA samples extracted from the biopsied muscle revealed a heteroplasmic m.10158T>C mutation, with a heteroplasmy level of 69.6%, in the mitochondrial complex I subunit geneMT-ND3. In contrast, this mutation was not found in her peripheral blood cells.
FINAL DIAGNOSIS
The diagnosis was MELAS syndrome harboring the m.10158T>C mutation.
TREATMENT
We administered levetiracetam (1000 mg/d) and oxcarbazepine (400 mg/d) to control epilepsy. Q10 and L-arginine were also administered.
OUTCOME AND FOLLOW-UP
Over the next 6 mo, the present patient did not manifest any further stroke-like episodes.
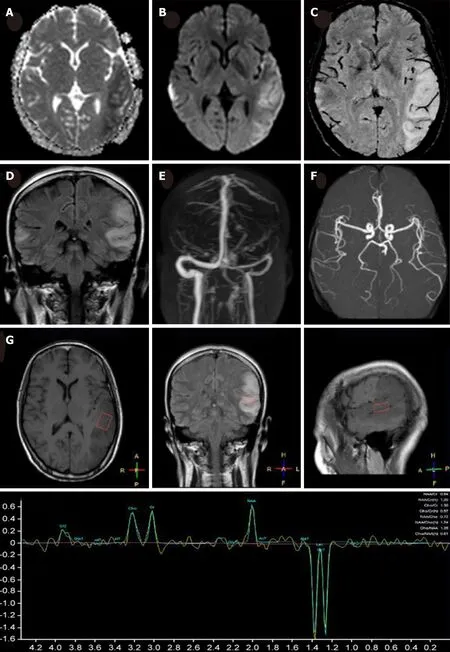
Figure 1 Magnetic resonance imaging of the present patient during her second hospitalization. Axial brain magnetic resonance imaging showed marked hyperintensity on diffusion-weighted imaging (A), slight hypointensity on apparent diffusion coefficient maps (B) in the bilateral temporal cortical regions, predominantly on the left side, with hyperintensity in the corresponding region on coronal fluid-attenuated inversion recovery (D). No hypointensity of dilated intrasulcal venous structures was observed on susceptibility weighted imaging (C), and no abnormalities were observed on magnetic resonance angiography (E) or magnetic resonance venography (F). Magnetic resonance spectroscopy revealed an inverted lactate (Lac) doublet in the subcortical region of the left temporal lesion (G).
DISCUSSION
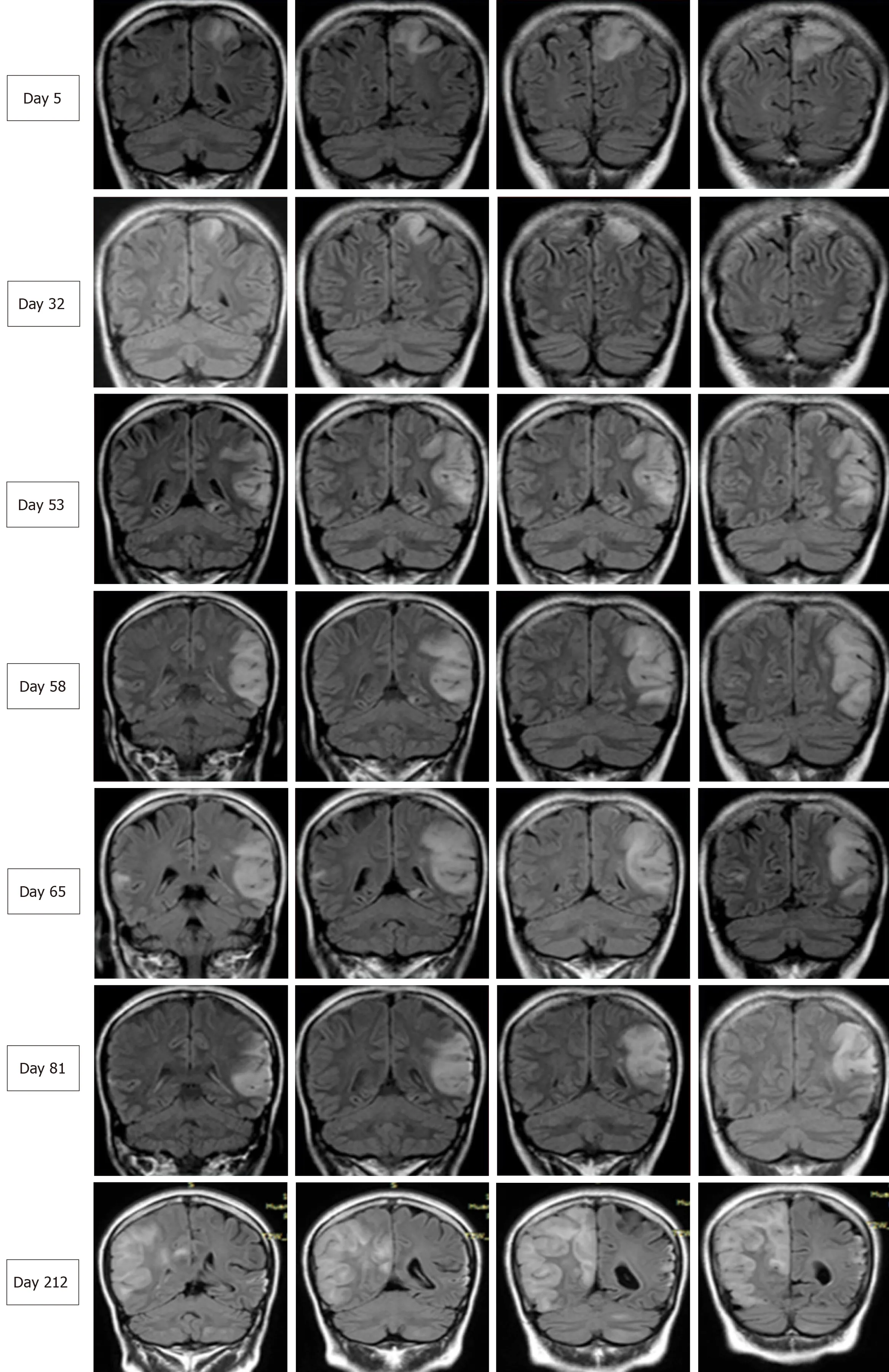
Figure 2 Serial brain images covering three recurrent stroke-like episodes in the present patient over a 7-mo period. Four representative coronal slices were serially shown in chronological order. Fluid-attenuated inversion recovery images on days 5, 32, 58, 65, 81, and 212 revealed hyperintensity signals appearing recurrently in various cortical and subcortical areas, mainly in the parietal and temporal lobes, and the progressive development of cortical atrophy.
We herein report a case of adult-onset mitochondrial encephalopathy, whereby the patient was admitted three times for similar stroke-like episodes before the definitive diagnosis was confirmed. Despite highly suggestive MRI features of MELAS syndrome, the diagnosis of mitochondrial encephalopathy was delayed due to the following reasons: (1) Lack of family history showing maternal inheritance; (2)Isolated symptoms of CNS impairment, without myopathy, lactic acidosis, or other systemic disorders; and (3) Negative mitochondrial genetic testing using peripheral blood. Although muscle biopsy revealed no abnormality, genetic testing using the biopsied muscle demonstrated a 69.6% heterozygous T10158C mutation in theMTND3gene.
Although MELAS is not uncommon in clinical practice, its diagnosis may be challenging because the clinical symptoms are highly variable and genetic findings may be absent. Our patient presented with a headache, seizure, and acute focal lesion observed by MRI, which fit the diagnostic criteria of supportive MELAS by Yatsugaet al[2]. Although her level of lactic acid in blood was normal, the presence of an inverted lactate peak was detected in the temporal cortex by MRS, which is a useful tool to detect metabolic dysfunction in the brain[3]. Additionally, despite no obvious myopathy, the biopsied muscle showed a 69.6% heteroplasmic mutation, T10158C, in theMT-ND3gene.
TheMT-ND3protein is a structural component of multimeric enzyme complex I of the mitochondrial respiratory chain, which drives ATP generation in mitochondria[4].TheMT-ND3T10158C mutation, which is located in the coding region of the loop domain of the ND3 subunit protein, has been identified in infants and pediatric patients with Leigh syndrome or Leigh-like disease[5]. However, the manifestation of late-onset stroke-like encephalopathy similar to MELAS in our patient harboring theMT-ND3T10158C mutation highlights the complicated genotype-phenotype relationship in mitochondrial diseases. In a literature review, we found four similar cases ofMT-ND3T10158C mutation (in muscle)[6,7], and all of these patients presented with adult-onset, isolated involvement of the central nervous system. Moreover, MRI revealed similar features among these five patients, with lesions predominately in the posterior cortex of the supratentorial region; in contrast, MRI typically shows involvement of the basal ganglia, brainstem, and cerebellum in Leigh syndrome.Thus, adult-onset patients harboring theMT-ND3T10158C mutation may exhibit unique clinical features. Although no myopathy symptoms were observed in any of these cases and genetic testing using peripheral blood was negative, muscle specimens should be obtained to establish a definitive diagnosis. This may be explained by heteroplasmic mtDNA mutations in various tissues, in which case, the mutation load in muscles is higher than that in blood[8].
CONCLUSION
Adult-onset mitochondrial encephalopathy in the presence of theMT-ND3T10158C mutation shows unique clinical characteristics. Despite a lack of lactic acidosis,myopathy, or other clinical implications, the diagnosis of MELAS needs to be considered for adult-onset patients with a stroke-like episode of encephalopathy. Due to the heteroplasmic mutation load, a negative genetic test obtained when using peripheral blood does not necessarily exclude the diagnosis of mitochondrial disease,and next-generation DNA sequencing using biopsied muscle should be considered.
 World Journal of Clinical Cases2019年9期
World Journal of Clinical Cases2019年9期
- World Journal of Clinical Cases的其它文章
- Coexistence of breakpoint cluster region-Abelson1 rearrangement and Janus kinase 2 V617F mutation in chronic myeloid leukemia: A case report
- Crizotinib-induced acute fatal liver failure in an Asian ALK-positive lung adenocarcinoma patient with liver metastasis: A case report
- Rare variant of pancreaticobiliary maljunction associated with pancreas divisum in a child diagnosed and treated by endoscopic retrograde cholangiopancreatography: A case report
- Nerve coblation for treatment of trigeminal neuralgia: A case report
- Management of the late effects of disconnected pancreatic duct syndrome: A case report
- Sofosbuvir/Ribavirin therapy for patients experiencing failure of ombitasvir/paritaprevir/ritonavir + ribavirin therapy: Two cases report and review of literature