
ARTICLE Vibrational and Structural Dynamics of Mn(CO)5Br and Re(CO)5Br Examined Using Nonlinear Infrared Spectroscopy?
2016-04-08 06:35MinjunFengFnYngJinpingWngBeijingNtionlLbortoryforMoleculrSciencesMoleculrRectionDynmicsLbortoryInstituteofChemistryChineseAcdemyofSciencesBeijing100190ChinbUniversityofChineseAcdemyofSciencesBeijing100049ChinDtedRecei
Min-jun Feng?,Fn Yng?,Jin-ping Wng?.Beijing Ntionl Lbortory for Moleculr Sciences,Moleculr Rection Dynmics Lbortory, Institute of Chemistry,Chinese Acdemy of Sciences,Beijing 100190,Chinb.University of Chinese Acdemy of Sciences,Beijing 100049,Chin(Dted:Received on December 15,2015;Accepted on Jnury 13,2016)
?
ARTICLE Vibrational and Structural Dynamics of Mn(CO)5Br and Re(CO)5Br Examined Using Nonlinear Infrared Spectroscopy?
Min-jun Fenga,b?,Fan Yanga?,Jian-ping Wanga?
a.Beijing National Laboratory for Molecular Sciences,Molecular Reaction Dynamics Laboratory, Institute of Chemistry,Chinese Academy of Sciences,Beijing 100190,China
b.University of Chinese Academy of Sciences,Beijing 100049,China
(Dated:Received on December 15,2015;Accepted on January 13,2016)
Vibrational and structural dynamics of two transition metal carbonyl complexes,Mn(CO)5Br and Re(CO)5Br were examined in DMSO,using ultrafast infrared pump-probe spectroscopy, steady-state linear infrared spectroscopy and quantum chemistry computations.Two carbonyl stretching vibrational modes(a low-frequency A1mode and two high-frequency degenerate E modes)were used as vibrational probes.Central metal e ff ect on the CO bond order and force constant was responsible for a larger E-A1frequency separation and a generally more red-shifted E and A1peaks in the Re complex than in the Mn complex.A generally broader spectral width for the A1mode than the E mode is believed to be partially due to vibrational lifetime e ff ect.Vibrational mode-dependent diagonal anharmonicity was observed in transient infrared spectra,with a generally smaller anharmonicity found for the E mode in both the Mn and Re complexes.
Key words:Transition metal carbonyl,Transient IR spectroscopy,Vibrational relaxation, Anharmonicity
?Part of the special issue for“the Chinese Chemical Society’s 14th National Chemical Dynamics Symposium”.
?They contributed equally to this work.
?Author to whom correspondence should be addressed.E-mail: jwang@iccas.ac.cn,Tel.:+86-10-62656806,FAX:+86-10-62563167
I.INTRODUCTION
It is well known that transition metal carbonyl coordination compounds play a very critical role in catalyzing chemical reactions,however,it still remains as a challenge for chemists to uncover the fundamental mechanisms lying underneath the complicated catalytical processes[1].Vibrational excited states,in particular those of carbonyl ligands,are very important in these catalytic reactions.Infrared(IR)spectroscopy,particularly ultrafast time-resolved IR spectroscopy,is a powerful tool to investigate complicated molecular structures and structural processes at the level of chemical-bond structural sensitivity and on the time scale of femtosecond temporal sensitivity[2?4]. Tabletop Ti:sapphire laser and optical parametric ampli fi er technologies can now generate stable femtosecond IR pulses,providing excellent light sources for ultrafast transient IR spectroscopic measurements.One of such measurements is pump-probe.In a typical IR pump-probe experiment,a relatively intense IR pulse (referred as pump)is used to excite a subensemble of a condensed-phase molecule(solute)to a speci fi c vibrational excited state,a weak IR pulse(referred as probe) is followed with a varying delay time to characterize the vibrational relaxations of the excited states.Because molecular vibrations are directly in fl uenced by the molecular structures as well as by instantaneous solvent con fi gurations,the IR pump-probe method provides a sensitive measurement of both vibrational relaxations and molecular structural dynamics.With the aid of femtosecond IR laser pulses,the time resolution of this method is readily comparable to that of molecular dynamics simulations.
Manganese and rhenium pentacarbonyl bromides (Mn(CO)5Br and Re(CO)5Br)are typical transition metal carbonyl coordination compounds that can be used as model molecules to understand the vibrational properties of these transition metal carbonyl species. The two complexes share the same formula,M(CO)5Br (M=Mn and Re respectively),and have C4vsymmetry.In this work,we are particularly concerned with the carbonyl stretching mode(νCO)whose vibrational frequency is located in the 5-μm wavelength region (2000 cm?1in wavenumber).The vibrational representation of M(CO)5Br complexes contains two infraredactive νCOmodes(A1and E)[5],which are mainly the axial and equatorial CO stretching vibrations respectively.While it has been established that the vibrational frequency of these two modes is in the order of E>A1,the vibrational and structural dynamics underneath these two modes,and the in fl uence of central metal atom on the vibrational dynamics,are less under-stood.
Further,the carbonyl stretching mode can be e ff ectively used as a vibrational probe for the structures and dynamics of the transitional metal carbonyls[6].The carbonyl stretching mode has a large transition dipole moment and an absorption linewidth that are sensitive to both molecular structure and solvent environment [7].The carbonyl stretching mode also usually has a vibrational lifetime that falls into the picosecond time regime[8],providing a time window for examining the ultrafast structural dynamics of these transition metal compounds.An aprotic polar solvent,namely dimethyl sulfoxide(DMSO),was selected in this work because it can dissolve manganese and rhenium pentacarbonyl bromides at reasonable concentration,also because it is relevant in catalytic and photophysical studies.
II.MATERIALS AND METHODS
A.Sample and FTIR experiments
Mn(CO)5Br and Re(CO)5Br were purchased from Sigma-Aldrich and used without further puri fi cation. The two transition metal carbonyl complexes were dissolved in DMSO at concentrations of 10.8 and 17.1 mmol/L,respectively.The sample solutions were loaded into a home-made liquid IR sample-cell,which is composed of two 2-mm thick calcium fl uoride windows and separated by a 50-μm thick Te fl on spacer.FTIR spectra were collected using a Nicolet 6700 FTIR spectrometer(Thermo Electron)at 1-cm?1spectral resolution.The FTIR experiments were carried out at room temperature(22?C).
B.Pump-probe spectroscopy
The transient IR pump-probe experiments were carried out with a home-built IR pump-probe spectrometer[9,10].Mid-infrared pulses with central frequency at near 2000 cm?1were generated via a di ff erence frequency generator(DFG)that was pumped by an optical parametric ampli fi er(OPA),and the latter was pumped by a Ti:sapphire laser ampli fi er system(sub 35-fs pulse width,800-nm central wavelength,and 1-kHz repetition rate).The pump and probe pulses were generated by a beam splitter subsequently collimated and focused on the sample cell by a focusing parabolic mirror with 10-cm focal length.The time delay between the pump and probe pulses was controlled with a motorized translation stage.The polarization direction of the probe pulse,with respect to that of the pump pulse,was adjusted to 54.7?(the so-called“magic”angle[11])via a half-wave plate and a polarizer,at which angle the lifetime of the vibrational excited state can be measured free from molecular reorientational e ff ects.
Transient spectra were obtained by chopping the pump pulse at half of the laser repetition.Sample position was controlled with a manual mechanical XY Z-translation stage.The nonlinear IR experiments were carried out at room temperature(22?C).
C.Quantum chemistry calculation
Geometry optimization of both Mn(CO)5Br and Re(CO)5Br was carried out and followed by subsequent vibrational frequency analysis including anharmonicity computations,in implicit solvent using polarizable continuum model(PCM)[12,13]with default parameters for DMSO.Density functional(B3LYP)with the 6-31++G??basis set for light atoms(C,O,and Br) and the Lanl2dz pseudopotential for the central transition mental atom were used.All computations were performed using the density functional theory(DFT) as implemented in Gaussian 09[14].
III.RESULTS AND DISCUSSION
A.Linear IR spectra
The infrared spectra of Mn(CO)5Br and Re(CO)5Br dissolved in DMSO in the CO stretching region are shown in Fig.1 with peak intensity normalized.Clearly, for each compound, fi ve CO groups exhibit only two absorption peaks,and show compound-dependent peak positions and peak separations.The bands from the high-to low-frequency direction are denoted as two infrared-active νCOmodes(E and A1)respectively by symmetry coordinate[5],which are mainly the axial and equatorial CO stretching vibrations respectively.It is noticed that the E mode is a doubly degenerate mode with identical frequency.This is veri fi ed by DFT computations(see below).
Table I is the fi tting result of the experimental IR spectra using Voigt function.The Voigt function is used because neither pure Gaussian nor pure Lorentzian function could yield a satisfactory fi tting result.Peak widths are reported in the form of full-width at halfmaximum(FWHM),showing a tendency of increase as the metal center changes from manganese to rhenium. In addition,for the same molecule,the FWHM for the E mode is generally narrower than that for the A1mode, whose signi fi cance will be explained in later section.
B.Computations

?
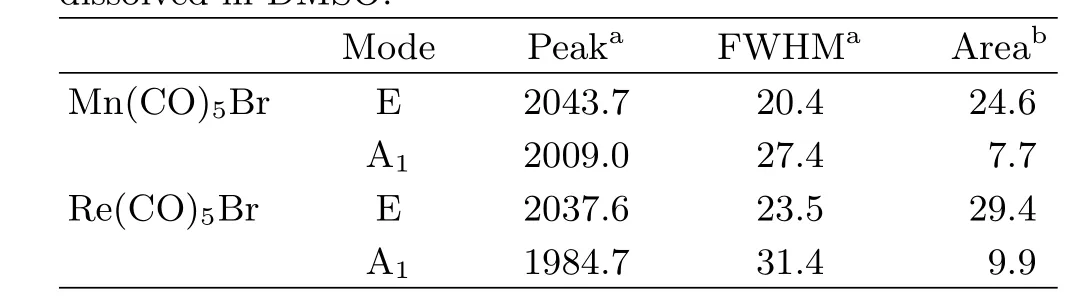
TABLE I Voigt function fi tting parameters of the linear IR spectra of the CO stretching of Mn(CO)5Br and Re(CO)5Br dissolved in DMSO.

FIG.1 Linear IR spectra of Mn(CO)5Br and Re(CO)5Br dissolved in DMSO in the CO stretching region.The bands from low frequency to high frequency in DMSO are denoted as A1and E.
To understand the observed IR spectra and vibrational frequency change associated with central atom, we performed quantum chemistry calculations on each complex in implicit DMSO solvent using the PCM method.Figure 2 shows the two optimized structuresaPeak position and FWHM are in unit of cm?1.bArea is in unit of arbitrary unit. with key bond distances listed,each showing a C4vsymmetry with relatively elongated CO bond length in the case of Re(CO)5Br.Table II lists the computed vibrational parameters.In Table II,the IR active A1and doubly degenerate E modes are predicted.Normalmode analysis shows that the E mode is mainly due to four radial CO stretching vibrations,while the A1mode is mainly due to the single axial CO stretching.This is in agreement with previous assignment[5].The computation not only reproduces experimentally observed order for the E and A1modes in both frequency and intensity in each complex,but also reproduces the frequency shift as a function of metal center mainly in two aspects: fi rst,as metal center goes from Mn to Re,both vibrational modes are red shifted(i.e.towards the lowfrequency side);second,the E and A1frequency separation becomes larger as metal center goes from Mn to Re.Because the two complexes are believed to have identical molecular symmetry(C4v)[15],these changes are attributed to the e ff ect of the central atom,which both belong to the VIIB group in the Periodic Table of element.

FIG.2 Molecular structure of Mn(CO)5Br and Re(CO)5Br with C4vsymmetry.These important bond lengths are marked in
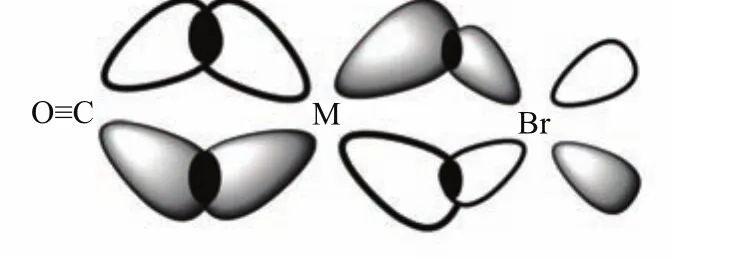
FIG.3 Carbon monoxide ligand acts as a strong acceptor in Mn(CO)5Br and Re(CO)5Br complexes.Only the axial direction is shown.
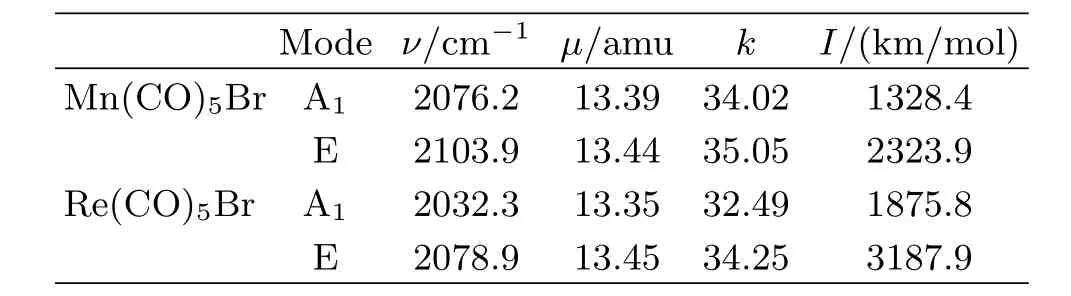
TABLE II Calculated CO stretching vibration frequency ν,reduced massμ,force constant k(in unit of mDyne/?A), transition intensity I for the A1and E modes in Mn(CO)5Br and Re(CO)5Br.
For carbonyl metal complexes,the intense carbonyl stretching bands are known to be closely related to back bonding e ff ects[16]that result from the interaction between the unoccupied antibonding(π?)orbitals of the carbonyl group and the d orbital electrons of the metal center.This is illustrated in Fig.3.Further,because the d orbital electron energy in Re is lower than that in Mn,a better energy match with the energy of theπ?orbitals of the carbonyl ligand in the case of Re is expected,thus forming stronger back bonding.Such a stronger back bonding also causes a Re?C bond length shortening in comparison with a single ReCO case(not given here).This causes C atom to be less positively charged and thus weakens the CO bond strength(decreases its bond order)and generally increases its bond length(Fig.2),resulting in a lowered vibrational frequency in the Re(CO)5Br complex than that in the Mn(CO)5Br complex.Because the four equatorial CO share the d orbital interaction,a rather limited red shift in frequency is expected for the E mode in the Re complex with respect to that in the Mn complex.This is the reason for larger E-A1frequency separation in the Re complex.
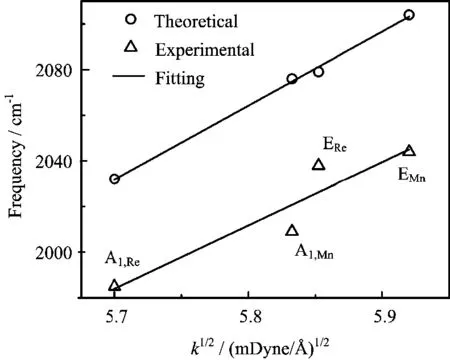
FIG.4 Theoretically computed frequency versus force constant for the CO stretching modes of Mn(CO)5Br and Re(CO)5Br.Experimentally observed frequencies are also plotted.
Further,Table II suggests that the metal atom e ff ect is mainly re fl ected in the force constant of the carbonyl stretch.Figure 4 shows the relationship between the computed frequency and the square root of the force constant for the E and A1modes in two transition metal complexes,which is indeed more or less linearly correlated as predicted.The experimental frequencies are also plotted for a direct comparison,in which a linear fi tting also seems reasonable.On the other hand,it should be pointed out that the reduced mass is also a factor because the frequency is proportional toμ?1/2. However,from the A1mode to E mode,their frequencies are not proportional toμ?1/2,because the two modes have slightly di ff erentμvalues.In addition,the CO bond lengths also generally increase in Re(CO)5Br as shown in Fig.2,which is the result of stronger back bonding.The increased bond length is apparently proportional to the increased frequency as well.
C.Pump-probe spectroscopy
The pump-probe spectra of both Mn(CO)5Br and Re(CO)5Br were obtained in the CO stretching vibration frequency region,of which the results are shown in Fig.5.This contour plot maps the state evolution of the vibrationally excited carbonyl stretching as a function of the pump-probe delay time.The magic-angle polarization condition is chosen so that the molecular orientational e ff ect is removed and only true vibrational relaxation can be seen.Here for simpli fi cation the zero delay time is set to when maximum pump-probe signal was observed,and instrumental response function deconvolution was not carried out.Nevertheless,the lifetime evaluation,which is found to be tens of picosecond(see below),will not be signi fi cantly a ff ected by the inclusion of the instrumental response function.Two types of signals corresponding to a given peak in the linear IR(e.g.the E mode),are shown as a negative signal (plotted in blue color)due to vibrational-state bleaching and stimulated emission(from v=0 to v=1,where v is vibrational quantum number),and as a positive signal(plotted in red color)due to the fi rst excited state absorption(from v=1 to v=2).The red and blue signals are separated in the frequency range shown in Fig.5 due to vibrational anharmonicity(?=δE0?1?δE1?2,where δE is corresponding two-level vibrational energy separation).

FIG.5 Magic-angle pump-probe spectra of(a)Mn(CO)5Br and(c)Re(CO)5Br in the CO stretching region.Decomposition of transient spectra at 0 fs delay time of(b)Mn(CO)5Br and(d)Re(CO)5Br for anharmonicity evaluation.
The E and A1modes are more separated in frequency for the Re complex than for the Mn complex,so that the absorption and bleach signals are clearly shown in the former but not in the latter.Note that ground-state bleaching signal of Mn(CO)5Br in the A1mode appearsto be missing in Fig.5(a)due to signal overlap between the 0-1 and 1-2 transitions.This missing spectral component can be identi fi ed by spectral fi tting(Fig.5(b)).

TABLE III Vibrational lifetime(T1)obtained by single exponential fi tting the magic-angle data for the Mn(CO)5Br and Re(CO)5Br complexes at desired frequency positions: bleaching(ν0→1)and absorption(ν1→2).
To have a detailed understanding of the ultrafast dynamics in relative to those vibrational transitions investigated,we extracted the temporal evolution of the population distribution at the maximum intensity of the excited state transitions,which are shown in Fig.6 for the Mn(CO)5Br and Re(CO)5Br complexes.These vibrational relaxation traces were fi tted reasonably by single exponential decay,of which parameters are summarized in Table III.
One may intuitively think that those two complexes should have the same vibrational relaxation behavior for their very much alike molecular structure.However,our experiment results show that there is a significant di ff erence between the T1times for the E and A1modes in a given transition metal complex,and there is also some dependence on the transition metal center. Speci fi cally,for the Mn complex,the T1time of the A1mode(87.7 ps)is nearly 10%faster than the E mode (ca.101 ps).For the Re complex,the T1time of the A1mode(ca.80 ps,averaged from v=0 to v=1 and v=1 to v=2 transitions)is also nearly 10%faster than the E mode(ca.90 ps,averaged from v=0 to v=1 and v=1 to v=2 transitions).
These T1values are found to roughly anticorrelate with the behavior of the linewidth shown in the linear IR spectra(Fig.1),that is,the shorter the T1,the broader the spectral width,which is found to be true for both Mn and Re complexes.In addition,the linewidth in the E and A1modes are generally broader in the Re complex than in Mn complex,which is also in agreement with relatively shorter T1observed in the Re complex than in Mn complex.Of course,the discussion here simply ignores the contribution of inhomogeneous broadening.The question is why the A1mode is nearly 10% shorter lived than the E mode?It could be due to a better energy match between the A1mode and linear combinations of low-frequency solvent modes;it could be also due to the axial bromide ligand e ff ect.Even though the data are solid,a de fi nite answer cannot be provided at this moment.
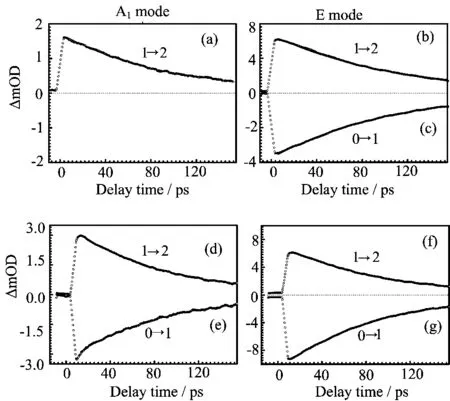
FIG.6 Population relaxation dynamics of the CO stretching mode of(a,b,and c)Mn(CO)5Br and(d,e,f,and g) Re(CO)5Br.Experimental data(circle)and exponential fi tting(solid line)are given.Probing frequency positions are marked as dashed lines in Fig.5(a)and(c).
D.Anharmonicity
In Fig.5 the contour plot clearly shows the presence of anharmonicity for both the E and A1modes in the two transition metal carbonyls.By taking a slice of the time-dependent transient absorption spectra at a given delay time,the anharmonicity of each mode can be extracted.Early-time spectra were used to avoid crosspeak contributions due to intramolecular vibrational redistribution that may occur at longer time[17].The results are shown in Fig.5(b)and(d).Here the diagonal anharmonicity for a given mode is measured,which is de fi ned as?=δE0?1?δE1?2,as mentioned above.For each transient spectrum,two pairs of Gaussian functions were utilized(for simpli fi cation)with negative peak positions fi xed to those observed in the linear IR spectra(Fig.1).For the E mode the anharmonicity was determined to be 14.3 cm?1,while for the A1mode the anharmonicity was 17.7 cm?1.For the Re complex,the anharmonicity of the E and A1mode was determined to be 13.3 and 24.9 cm?1,respectively.
To evaluate the diagonal anharmonicity of the carbonyl stretching modes in the two transition metal complex,ab initio computations were carried out using the method outlined in previous work[18,19].It was found that for both the Mn and Re complexes,the diagonal anharmonicity of the E mode is generally smaller than that of the A1mode(data not shown),which is in general agreement with the experimental results.
These results show that due to the nature of thevibrational mode(A1mode is mainly the axial CO stretching vibration while the degenerate E modes are mainly the linear combination of the four equatorial CO stretching vibrations),the diagonal anharmonicity of the E modes is generally smaller than that of the A1mode.This is understandable because more delocalized modes tend to smaller diagonal anharmonicity [19],which is the case of E mode in the present work. No obvious metal center dependence can be obtained, either due to the limitation of the spectral-resolution of the pump-probe method in measuring the anharmonicity,or due to the possibility that the metal-center dependence on the carbonyl stretching anharmonicity is insigni fi cant for the two cases studied here.At the moment,our data cannot di ff erentiate these two possibilities.
IV.CONCLUSION
In this work,we investigated two typical transition metal carbonyl coordination compounds which have different central metal atoms,namely Mn(CO)5Br and Re(CO)5Br.Examination of the carbonyl stretching modes of the two compounds in an aprotic but polar solvent(DMSO)using steady-state linear infrared and time-resolved nonlinear infrared methods yields detailed insights into the structural and vibrational relaxation dynamics of the two carbonyl complexes in solution phase.From Mn to Re,the metal center dependent linear IR feature is seen and explained as the π backbonding e ff ect,which results in a larger E-A1mode vibrational frequency separation and a generally red-shifted E and A1modes in the case of Re.A generally broader spectral width for the low-frequency A1mode than the high-frequency E mode is believed to be partially due to the vibrational lifetime(T1)e ff ect.By ignoring the inhomogeneous broadening contribution, the T1time is shorter for the A1mode,whose spectral width(FWHM)is also broader,due to the well-known 1/2T1contribution to the total spectral linewidth[2]. This is found to be true in both Mn and Re complexes. Finally vibrational diagonal anharmonicity for the E and A1modes are measured from the early-time transient pump-probe spectra.The anharmonicity of the E mode is found to be generally smaller in both Mn and Re complexes,which is believed to be associated with the more delocalized nature of the E mode.These results provide fundamental measurements of the vibrational signatures of the VIIB-group transition metal carbonyl complexes.
V.ACKNOWLEDGMENTS
This work was supported by the Hundred Talent Fund of the Chinese Academy of Sciences,and also supported by the National Natural Science Foundation of China(No.21473212,No.20727001 and No.21573243). The author thanks P.Yu and J.Zhao for their technical assistances.
[1]M.Chergui,Acc.Chem.Res.48,801(2015).
[2]J.C.Owrutsky,D.Raftery,and R.M.Hochstrasser, Annu.Rev.Phys.Chem.45,519(1994).
[3]M.D.Fayer,Annu.Rev.Phys.Chem.21(2009).
[4]L.M.Kiefer,J.T.King,and K.J.Kubarych,Acc. Chem.Res.48,1123(2015).
[5]D.Kariuki and S.F.A.Kettle,Spectrochim.Acta A 34,563(1978).
[6]J.K.McCusker,Acc.Chem.Res.36,876(2003).
[7]E.J.Heilweil,R.R.Cavanagh,and J.C.Stephenson, Chem.Phys.Lett.134,181(1987).
[8]E.J.Heilweil,R.R.Cavanagh,and J.C.Stephenson, J.Chem.Phys.89,230(1988).
[9]D.Li,F.Yang,C.Han,J.Zhao,and J.Wang,J.Phys. Chem.Lett.3,3665(2012).
[10]F.Yang,P.Yu,J.Zhao,and J.Wang,ChemPhysChem 14,2497(2013).
[11]G.Fleming,Chemical Applications of Ultrafast Spectroscopy,1st Edn.,Oxford:Oxford University Press (1986).
[12]E.Canc`es,B.Mennucci,and J.Tomasi,J.Chem.Phys. 107,3032(1997).
[13]B.Mennucci and J.Tomasi,J.Chem.Phys.106,5151 (1997).
[14]M.J.Frisch,G.W.Trucks,H.B.Schlegel,G.E. Scuseria,M.A.Robb,J.R.Cheeseman,G.Scalmani, V.Barone,B.Mennucci,G.A.Petersson,H.Nakatsuji,M.Caricato,X.Li,H.P.Hratchian,A.F.Izmaylov,J.Bloino,G.Zheng,J.L.Sonnenberg,M. Hada,M.Ehara,K.Toyota,R.Fukuda,J.Hasegawa, M.Ishida,T.Nakajima,Y.Honda,O.Kitao,H.Nakai, T.Vreven,J.A.Montgomery Jr.,J.E.Peralta,F. Ogliaro,M.Bearpark,J.J.Heyd,E.Brothers,K.N. Kudin,V.N.Staroverov,R.Kobayashi,J.Normand, K.Raghavachari,A.Rendell,J.C.Burant,S.S.Iyengar,J.Tomasi,M.Cossi,N.Rega,J.M.Millam,M. Klene,J.E.Knox,J.B.Cross,V.Bakken,C.Adamo, J.Jaramillo,R.Gomperts,R.E.Stratmann,O.Yazyev, A.J.Austin,R.Cammi,C.Pomelli,J.W.Ochterski, R.L.Martin,K.Morokuma,V.G.Zakrzewski,G.A. Voth,P.Salvador,J.J.Dannenberg,S.Dapprich,A. D.Daniels,¨O.Farkas,J.B.Foresman,J.V.Ortiz,J. Cioslowski and D.J.Fox,Gaussian 09,Revision A.02, Wallingford,CT:Gaussian Inc.,(2009).
[15]Y.F.Hu,G.M.Bancroft,and K.H.Tan,Inorg.Chem. 39,1255(2000).
[16]G.L.Miessler,P.J.Fischer,and D.A.Tarr,Inorganic Chemistry,5th Edn.,New Jersey:Pearson(2013).
[17]F.Yang,P.Yu,J.Zhao,J.Shi,and J.Wang,Phys. Chem.Chem.Phys.17,14542(2015).
[18]V.Barone,J.Chem.Phys.122,014108/1(2005).
[19]J.Wang and R.M.Hochstrasser,J.Phys.Chem.B 110,3798(2006).
 CHINESE JOURNAL OF CHEMICAL PHYSICS2016年1期
CHINESE JOURNAL OF CHEMICAL PHYSICS2016年1期
- CHINESE JOURNAL OF CHEMICAL PHYSICS的其它文章
- ARTICLE E ffi cient Separation of Ar and Kr from Environmental Samples for Trace Radioactive Noble Gas Detection?
- REVIEW Polarization Dependent Time-Resolved Infrared Spectroscopy and Its Applications?
- ARTICLE Reactions of Group V Metal Atoms with Hydrogen Sul fi de:Argon Matrix Infrared Spectra and Theoretical Calculations?
- ARTICLE Structural Dynamics of Phenyl Azide in Light-Absorbing Excited States: Resonance Raman and Quantum Mechanical Calculation Study?
- ARTICLE Structural and Infrared Spectroscopic Study on Solvation of Acetylene by Protonated Water Molecules?
- ARTICLE Excited-State Proton Transfer and Decay in Hydrogen-Bonded Oxazole System:MS-CASPT2//CASSCF Study?