
Interstitial Mycosis Fungoides with Systemic Sclerosis-Like Features: A Case Report
2019-10-16 11:05:14QiuJuMiaoYiFeiWangYiGengXiuLianXu
國(guó)際皮膚性病學(xué)雜志 2019年3期
Qiu-Ju Miao, Yi-Fei Wang, Yi Geng, Xiu-Lian Xu?
Department of Pathology, Hospital for Skin Diseases (Institute of Dermatology), Chinese Academy of Medical Sciences and Peking Union Medical College, Nanjing, Jiangsu 210042, China.
Introduction
Mycosis fungoides (MF) is the most common form of cutaneous lymphoma, accounting for almost 50% of all types.1MF occasionally manifests as the interstitial lymphocytic infiltrate that mimics inflammatory morphea,interstitial granulomatous dermatitis, and interstitial granuloma annulare, and this rare histopathological variant has been termed interstitial mycosis fungoides(IMF). IMF can be considered as a transient histopathological pattern in conventional MF,with common clinical presentation as patches and plaques.2
Scleroderma refers to an autoimmune connective tissue disorders,including three subtypes:localized scleroderma(LSc or morphea), systemic sclerosis (SSc), and diffuse cutaneous systemic sclerosis.Cases with features clinically corresponding to localized scleroderma but histopathologically compatible with IMF have emphasized the importance of clinicopathological correlation between T-cell lymphoma and scleroderma.3There are also studies revealing the necessity of suspecting cutaneous T-cell lymphoma in view of prominent subcutaneous lesions in patients with rheumatic diseases.4However, cases of SSc-like MF or coexistence of MF and SSc have been rarely reported.Herein we report a case that a 70-year-old male with a 20-year history of psoriasis presented with progressive skin sclerosis, and was at first clinically diagnosed as SSc but turned out to be IMF after perfecting interrelated tests.
Case report
A 70-year-old Chinese man recently presented to the Hospital for Skin Diseases with a one-year history of dermal sclerosis on the trunk and limbs. One year previously, the skin of his trunk had become hard and dark with no obvious inducing factor. The symptoms gradually spread to all four limbs and progressed until his physical activity became limited. No other abnormalities,such as Raynaud’s phenomenon, gastrointestinal symptoms, or systemic symptoms, were detected at that time,and no treatments were given. He also had a history of psoriasis that was first diagnosed in our hospital 20 years ago and for which he did not receive regular treatments.He had no family history of similar diseases.
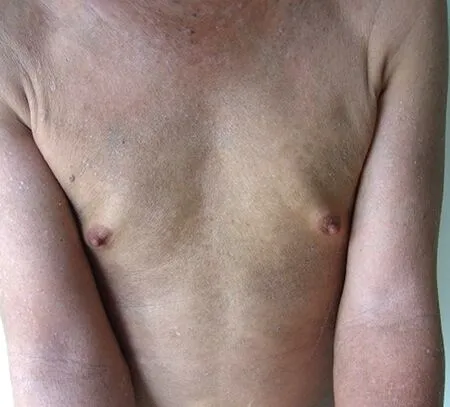
Figure 1. Clinical characteristics of the patient.Diffuse thickening and hardening of the skin on the trunk and all four limbs.
Physical examination revealed no enlarged superficial lymph nodes or other abnormalities. Dermatological examination revealed thickening, diffuse hardening, and pigmentation of the skin on the trunk and all four limbs;however,both hands were spared(Figure 1).Additionally,typical psoriatic lesions were seen on the inner surfaces of the limbs and scalp with a positive Auspitz sign.Laboratory analyses showed a normal lymphocyte count in the peripheral blood,and relevant antibody tests for SSc were negative.Biopsy specimens of the abdominal sclerotic lesions as examined by hematoxylin-eosin staining revealed a roughly normal epidermis, mild hyperplasia of the collagen fibers in the dermis, and infiltration of atypical lymphoid cells, some of them containing large,hyperchromatic nuclei. Mitosis was also observed in scattered cells without typical epidermotropism or Pautrier microabscess formation(Figure 2A).Immunohistochemical analysesshowedstrongexpressionofLCA,CD3,CD45RO,CD5,and CD4;40%Ki67 positivity;and CD8,CD20,and CD79a negativity (Figure 2B-2F). Monoclonal rearrangement of T-cell receptor genes was detected.
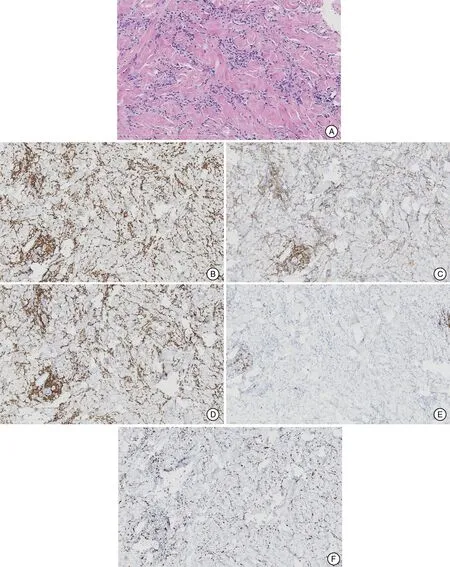
Figure 2. Histopathological analyses of the lesion.A:Hematoxylin and eosin staining revealed roughly normal epidermis with mild hyperplasia of the collagen fibers in the dermis; infiltration of atypical lymphoid cells, some with large, hyperchromatic nuclei; and mitosis in scattered cells(×200).B:CD3 positivity(×100).C:CD4 positivity(×100).D:CD5 positivity(×100).E:CD20 negativity(×100).F:40%Ki67 positivity(×100).
The patient was finally diagnosed as Stage III IMF,and then received CHOP chemotherapy in Jiangsu Cancer Hospital (cyclophosphamide, doxorubicin, vincristine,and prednisone; dose were unknown). However, the patient discontinued treatment and was eventually lost to follow-up.
Discussion
IMF is a rare histopathological variant of MF that typically manifests as an interstitial lymphocytic infiltrate in the dermis.MF is an indolent cutaneous T-cell lymphoma with a natural progression that is subdivided into four stages:patch, plaque, tumor, and visceral involvement. Apart from the classic presentations of patches and plaques,which should be regularly distinguished from interstitial granuloma annulare, inflammatory morphea, and interstitial granulomatous dermatitis, some unusual clinical features can make the diagnosis of MF much more difficult.2Accurate clinicopathological correlation and phenotypic studies of atypical dermal interstitial lymphohistiocytic infiltrates help to achieve the correct diagnosis.
In the present case, IMF clinically presented as diffuse thickening,hardening,and pigmentation of the skin on the trunk and all four limbs. Histological examination revealed long, linear aggregates of dermal lymphocytes splaying the collagen fibers,with very few plasma cells and no dermal mucin. Immunohistochemical examination confirmed that T cells predominated over histiocytes and B cells and that monoclonal rearrangement of T-cell receptor genes had occurred, favoring the diagnosis of IMF.However,theunusualSSc-likeclinicalfeaturesdeserve more attention since several cases revealing the correlation between scleroderma and MF have been reported.3,5
SSc is an autoimmune connective tissue fibrosing disease characterized by Raynaud phenomenon(95%),cutaneous sclerosis(75%),nail fold and fingernail alterations(80%),telangiectasia(75%),and skin thickening of the fingers of both hands extending proximal to the metacarpophalangeal joints, which is sufficient for the diagnosis alone.6In the present case, both of the patient’s hands were spared,and Raynaud phenomenon was absent. After further consideration of the negativity for specific autoantibodies,the differential diagnosis of SSc was finally ruled out, as were other mimics, including immune-mediated/inflammatory, genetic, drug-induced/toxic, metabolic, panniculitides/vascular, and (para) neoplastic disorders.
Skin-directed therapies are still widely used, especially appropriate for early-stage MF, including high-potency topical corticosteroids,topical mechlorethamine(0.02%),topical bexarotene (1%), ultraviolet phototherapy(8-methoxypsoralen plus 320-400nm ultraviolet A,ultraviolet B), total skin electron beam therapy (a total dose of 30-36 Gy applied over a period of 8-10 weeks),and localized radiotherapy (dose ranging from 0.7 to 35 Gy and may be fractionated). Systemic therapies,sometimes combined with skin-directed therapies, are recommended when equal to or greater than III stage.Interferon-alpha and retinoids,as moderate response rates achieved in monotherapy, are commonly used with plus ultraviolet A. Chemotherapy is conventional but still important for aggressive diseases covering CHOP, chlorambucil,methotrexate,and some novel chemotherapeutic agents such as pegylated liposomal doxorubicin and gemcitabine. Targeted immunotherapy, extracorporeal photochemotherapy,hematopoietic stem cell transplantation, and histone deacetylase inhibitors could also be considered.7In the present case, the patient was detected with lymphatic involvement in the further examination and was recommended chemotherapy but the treatment was soon interrupted regrettably.
Overall,high sensitivity and accuracy are very important during the diagnostic workup given that less than onethird of patients with MF develop an advanced stage and that a delayed diagnosis is a significant risk factor for advanced disease.
Acknowledgement
This study was supported by CAMS Innovation Fund for Medical Sciences (No. CIFMS-2017-I2M-1-107).

- 國(guó)際皮膚性病學(xué)雜志的其它文章
- Solid-Cystic Hidradenoma
- Arteriosclerosis Obliterans Presenting as Multiple Leg Ulcers: A Case Report
- Nasal Type Extranodal NK/T-Cell Lymphoma Presenting with Unilateral Facial Erythemas,Nodules, and Necrosis
- Lentigines within Nevus Depigmentosus
- Subungual Exostosis Misdiagnosed as Subungual Wart
- Severe Port Wine Stain with Significant Nodules and Alveolar Bone Invasion Leading to Restricted Mouth Opening