
Molecular mapping and candidate gene analysis of the semi-dominant gene Vestigial glume1 in maize
2019-11-12 08:29:36ChoxinLiuYunzengZhoYngBiXiominLuWeibinSongLipingQinYilinCi
The Crop Journal 2019年5期
Choxin Liu,Yunzeng Zho, Yng Bi, Xiomin Lu, Weibin Song, Liping Qin,Yilin Ci,*
aMaize Research Institute,Southwest University, Chongqing 400715, China
bHenan Institute of Science and Technology, Xinxiang 453003,Henan,China
cHenan Academy of Agricultural Sciences,Zhengzhou 450002,Henan,China
dNational Maize Improvement Center of China,China Agricultural University, Beijing 100193,China
Keywords:Vestigial glume1 Fine mapping Candidate gene analysis ZmCPA-FAS1 Glume development
ABSTRACT The glume is an organ of the maize spikelet and plays important roles in anther and kernel development. Vestigial glume1 (Vg1) is a classic mutant associated with ligule and glume development. Here we report the phenotypic characterization, fine mapping, and candidate gene analysis of the Vg1 mutant.Vg1 is a semi-dominant and pleiotropic gene,and also affects plant height,ear height,and tassel length. Vg1 ligule degeneration begins at the first leaf,and the Vg1 tassel and ear can be distinguished from those of wild-type plants when their lengths reach respectively 55 mm and 51 mm. Using a BC3 mapping population of 11,445 plants, we delimited the Vg1 functional site to an interval of 7.4 kb, flanked by the markers InDelLM and CRM6. A putative cyclopropane fatty-acid synthase gene (ZmCPA-FAS1) was hypothesized to underlie the mutant phenotype.We detected a Helitron insertion in the sixth intron of ZmCPAFAS1. Its presence caused abnormal alternative splicing of ZmCPA-FAS1 that conferred new characteristics on the Vg1 mutant. These findings are a basis for further discovery of the molecular mechanism underlying glume development and a potential guide for maize breeding of small-glume varieties,especially sweet corn breeding.
1. Introduction
Maize is a monoecious species with unisexual male inflorescences at the apex of the plant and female inflorescences several nodes below the tassel [1]. Maize inflorescences consist of a main spike and branches(except for the ear) covered with spikelets, each composed of a pair of leaflike organs called glumes that play important roles in anther and kernel development. From teosinte, the wild progenitor of maize, to modern cultivars, the evolution of soft glumes is arguably the most important event in the domestication history of maize [2]. It increased the suitability of the crop for human planting and harvesting [3].Thus, elucidating the molecular basis underlying glume development is critical for understanding the evolution of maize.
Unlike teosinte,which has a cupule and hardened glumes in the kernel structure, modern maize cultivars are more easily harvested owing to their softer and thinner glumes and naked kernels on the ear (with cupule collapsed). This change in fruit case morphology was a key event during maize domestication, and its genetic basis has been studied for decades. In 1993, Doebley et al. [2] mapped a major Mendelian locus on chromosome 4 using an F2population from a cross between maize and teosinte. Subsequently,tga1,which encodes a SQUAMOSA promoter-binding protein transcription factor,was identified to be the key locus in this domestication process [4]. A single amino-acid change within this gene underlies the phenotypic difference between teosinte and maize. As a result, the accessibility of kernels for harvest is greatly increased [5]. Pod corn,resembling those of most grasses, was once considered to be the ancestor of modern cultivated maize [6]. Its kernels are entirely enclosed in conspicuous long glumes. A genetic analysis revealed that this trait is controlled by a single dominant gene, Tu1 [7], with a compound locus in which a cis-regulatory mutation and gene duplication of Zmm19 lead to ectopic expression in the inflorescences [8]. Besides affecting glume development, Tu1 is also involved in multiple processes in maize, including phase transition,branch meristem formation, spikelet initiation, and sex determination [9], suggesting its pleiotropic effects. Although the functions of the tga1 and Tu1 genes have been deciphered, the mechanism underlying maize glume development remains unclear.
Vestigial glume1 (Vg1) is a spontaneous mutant discovered by Sprague [10] in 1931 that produces defects during the development of ligules and glumes. In contrast to those of Tu1, Vg1 glumes are greatly reduced, with only a stubby base remaining intact, resulting in the anther’s being almost completely exposed. The Vg1 mutation also affects ligule development, leading to the liguleless phenotype of maize.Vg1 is reported to be located in bin 1.07 on chromosome 1,flanked by TIDP5507 and IDP856 (https://maizegdb.org/data_center/locus/?id=12730). However, no further studies have been reported to date.
In the present study, we characterized the phenotype of the Vg1 mutant and investigated its pleiotropic effects on plant architecture. Using a map-based cloning strategy, we delimited Vg1 to a narrow interval,determined the candidate gene, and also examined its expression pattern. Our results will be useful for future research on the functional analysis of Vg1 and for further understanding the mechanisms underlying the development of maize glumes.
2. Materials and methods
2.1. Plant materials
The Vg1 mutant with Hi27 genetic background was obtained from the Maize Genetics Cooperation Stock Center (http://maizecoop.cropsci.uiuc.edu/)and crossed with the B73 inbred line. Plants with mutant phenotypes were repeatedly backcrossed to B73 to generate a series of backcross populations. The BC2population was used for genetic pattern analysis, and the BC3and BC5were used for Vg1 mapping and phenotypic observation, respectively. During the development of backcross populations, a few Vg1 mutants that produced normal pollen were successfully self-fertilized to generate BC3F2and BC3F3populations for agronomic trait evaluation and phenotypic observation. The backgrounds of some figures were processed by Adobe Photoshop (Adobe Systems, MA,USA).
2.2. Ligule observation
The seedlings of the BC5population were grown in a climate chamber with 16 h daylight at 28 °C and 8 h dark at 22 °C.When the second leaf was fully expanded, the first and second leaves were dissected from the Vg/+ plants and their wild-type siblings and observed under a stereoscope (Olympus SZ61,Tokyo,Japan)to determine the developmental stage at which differentiation of mutant from normal plants occurred.
2.3. Observation of glumes of the immature tassel and ear
The BC5plants were grown in an experimental field for about 30 days. Immature tassels and ears were dissected from the Vg/+ plants and wild-type siblings every three days and immediately observed under a scanning electron microscope(SEM,Hitachi SU3500,Tokyo,Japan)to determine the stage at which differences between mutant and normal plants emerged during glume development in the immature tassel and ear.
2.4. Agronomic trait evaluation
The BC3F2and BC3F3populations were used for agronomic trait evaluation in plants with Vg/+,Vg/Vg,and+/+genotypes.Plant height, ear height, tassel length, node number, and tassel branch number were measured.Statistical analysis was performed using the Data Processing System (DPS) software package[11].
2.5. Development of codominant molecular markers
The maize genomic sequence in the vicinity of the Vg1 gene was downloaded from Gramene(http://ensembl.gramene.org/Zea_mays/Info/Index?db=core). Simple sequence repeats(SSRs) were searched for using SSRHunter 1.3 [12], and then subjected to a BLAST search against the maize genomic sequences available in the National Center for Biotechnology Information database (NCBI; https://www.ncbi.nlm.nih.gov/).Single-copy SSRs were used as templates for designing primers with Primer-BLAST (http://www.ncbi.nlm.nih.gov/tools/primer-blast/Index.cgi?LINK_LOC=BlastHome).
To develop cleaved amplified polymorphic sequence(CAPS) markers, single-copy DNA fragments around the Vg1 locus were identified and sequenced. A multiple sequence alignment was performed with DNAMAN (https://www.lynnon.com/). DNA sequences of the Vg1/Vg1 and wild-type plants with single-nucleotide polymorphisms (SNPs) were used to develop CAPS markers using the SNP analysis tool SNP2CAPS[13].
2.6. Fosmid library screening and thermal asymmetric interlaced PCR (TAIL-PCR)
To construct a genomic fosmid library for the Vg/Vg plants,the CopyControl HTP Fosmid Library Production Kit(Epicentre, WI, USA) was used. The methods used for DNA fragment preparation, library titer determination, fosmid stability analysis, and genomic library screening followed those described by a previous study [14]. The positive fosmid clone was digested with EcoR I and Hind III(Thermo Scientific,MA, USA), and a DNA fragment of approximately 3 kb was recovered and subcloned into the plasmid vector pMD18-T(Takara, Shiga, Japan) for identifying the target fragment using gene-specific primer I-8 (forward primer: 5′-ATTCTGGCCGGGGGAGATGGTTT-3′, reverse primer: 5′-AGAACTGGGGGTTGTGAACCCGT-3′). TAIL-PCR was performed to isolate the flanking sequences of the mutant locus. Degenerate primers were used as previously described[15]. Gene-specific primers were designed referring the B73 genome using Primer-BLAST (Table S1). The resulting sequence was subjected to a BLAST search against the maize genomic database (http://ensembl.gramene.org/Zea_mays/Tools/Blast?db=core;r=1:101101009-101101036;
tl=d0fvEWZ0y49JQEgx-48616-14920544) and maize transposable element database (http://maizetedb.org/~maize/) for further analysis.
2.7. RNA extraction and first-strand cDNA synthesis
Maize tissues were dissected and immediately frozen at-80 °C. Approximately 0.2 g of each sample, ground in liquid nitrogen, was used for total RNA extraction using the RNAprep Pure Plant Kit (TIANGEN, Beijing, China). The total RNA was treated with DNase I (TIANGEN) to remove genomic DNA.Approximately 1 μg of total RNA was used as a template for first strand cDNA synthesis according to the specifications of the RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific). For retrieving full-length cDNA, the SMARTer RACE cDNA Amplification Kit(Clontech,CA,USA)was used according to the manufacturer’s instructions to synthesize 5′- and 3′-RACE Ready cDNA.
2.8. Quantitative RT-PCR (qRT-PCR) and semi-quantitative RT-PCR
qRT-PCR was performed using a CFX96 Touch Real-time PCR Detection System (Bio-Rad, CA, USA) with the THUNDERBIRD SYBR qPCR Master Mix (TOYOBO,Osaka,Japan)according to the manufacturer’s instructions.The PCR protocol was as follows: initial denaturation at 95 °C for 15 s, followed by 45 cycles of annealing and extension at 64 °C for 60 s. Each cDNA sample had three replicates. The actin gene (forward primer: 5′-TCACCCTGTGCTGCTGACCG-3′, reverse primer: 5′-GAACCGTGTGGCTCACACCA-3′) was used as an internal control. Semi-quantitative RT-PCR was performed as previously described [16]. The PCR protocol was as follows:initial denaturation at 94 °C for 30 s, followed by 26 cycles of annealing at 60 °C for 20 s and extension at 72 °C for 30 s or 60 s.
3. Results
3.1. Phenotypes and genetic pattern analysis of Vg1
The ligules of Vg1 plants diminished with only intact base remained(Fig.1-A,B).The tassel glumes were reduced to onethird of normal length, even to the size of vestigial glumes,leaving the anthers almost completely exposed (Fig. 1-C-F).Without the protection of glumes, the Vg1 anthers withered quickly under high temperature,except that a few plants shed viable pollen.The ear glumes of Vg1 plants were also severely shrunken,to near-invisibility on the exposed cob(Fig.1-G,H).The Vg11 gene is associated with flower development, given that lemmas and paleae were absent in Vg1 plants(Fig.1-E,F).
Vg1 was introduced into the B73 background by successive backcrossing to generate a series of backcross populations.The BC2population,composed of 1111 plants,segregated with 525 mutants and 586 wild-type siblings, fitting the 1:1 Mendelian ratio (χ2= 3.244, χ20.05= 3.84). This result confirmed that a single gene governs the Vg1 genotype.
3.2.Stages at which Vg1 ligule and glume defects were visible
When the second leaf was fully expanded, the first and second leaves of the mutant and its wild-type siblings were examined.As shown in Fig.2-A,at the junction of the sheath and blade, the wild-type plant showed a transparent and membrane-like structure (indicated by arrows) known as the ligule. However, the ligules of the first leaf of the Vg/+ plants were greatly diminished (Fig. 2-B), with only vestiges intact.Thus, ligule degeneration occurred in the early development stage of seedling growth.
To examine Vg1 glume development, immature tassels at different development stages were dissected for SEM observation.When the tassel lengths were 18 mm and 32 mm,the Vg/+ glumes developed normally, except that their growth was slightly delayed compared with that in the wild-type plant (Fig. S1-A-D). However, when the tassel length was 55 mm, it could be clearly observed that some anthers were exposed owing to rudimentary glumes(Fig.2-C, D).
As shown in Fig. S1-E-H, no differences were found when the lengths of the immature ear of the wild-type and Vg/+plants were 25 mm and 45 mm. When the ear reached 51 mm, it was clearly seen that the Vg/+ glume began to diminish (Fig. 2-E, F), resulting in a naked ovary and finally exposing the cob after kernel shedding.
3.3. Vg1 affects plant architecture

Fig.1-The Vg1 mutant phenotype.The ligules(A and B)and glumes of the tassel(C and D),cob(G and H)and spikelets(E and F)were observed (indicated by red arrows).The ligules and glumes are greatly reduced and lemmas and paleae absent in Vg1 plants.The backgrounds of panels A,B,G,and H were processed with image software.Solid black lines represent scale bars.WT,wild type.
In addition to its influence on the development of ligules and glumes,Vg1 affected other agronomic traits.In the BC3F2and BC3F3populations, Vg1/Vg1 mutant plants were shorter and displayed other abnormal phenotypes than the Vg1/+ mutant and its wild-type siblings (Fig. S2). Plant height, ear height, node number, tassel length, and tassel branch number were evaluated in these two segregating populations.Over 30 plants of each genotype in the BC3F2and BC3F3populations were analyzed. In comparison with Vg1/+plants, plant height, ear height, node number, and tassel length in Vg1/Vg1 plants were significantly reduced to 40.3%,39.9%,94.1%,and 80.5%,respectively.In comparison with the wild-type siblings, these traits in the Vg1/+ plants were reduced to 85.6%, 85.8%, 94.4%, and 84.6%, respectively.However, the tassel branch number of the Vg1/Vg1 plants was higher than that of the Vg1/+and wild-type siblings(Fig.3 column A).Similar phenomena were observed in the BC3F3population (Fig. 3 column B). All traits but node number and tassel branch number differed between three genotypes(P <0.01). Thus, Vg1 is a semi-dominant gene and strongly affects the architecture of the maize plant.
3.4. Map-based cloning of Vg1
Vg1 was previously mapped to bin 1.07 on chromosome 1.Thirty-one SSR markers in bin 1.07 were used to search for linked markers in two DNA pools (Table S2), which consisted of 10 Vg1/+and wild-type plants of the BC3population,leading to the identification of the Vg1-linked SSR markers umc1128 and bnlg1556. In 375 plants, 67 with crossovers between bnlg1556 and umc1128 were identified (32 recombinants identified by bnlg1556 and 35 identified by umc1128). The newly developed SSR, CAPS, and InDel markers (Table S3)were used to identify respectively 12 and five plants with crossovers between LM1 and RM1 and between LM2 and RM2,respectively. Thus, the Vg1 locus was initially mapped to an interval of approximately 2.5 Mb flanked by LM2 and RM2(Fig.4-A).
For further mapping of the Vg1 locus, we screened for recombinants in a large BC3population of 11,070 plants. We identified 148,88,and 12 plants with crossovers between LM2 and RM2,LM3 and RM3,and LM4 and RM4,respectively(Fig.4-B).We used three CAPS markers(CLM5,CRM5,and CRM6)and one InDel marker (InDelLM) to identify plants with crossovers between LM4 and RM4.Finally,we identified one recombinant at both CLM5 and InDelLM and two recombinants at each of CRM5 and CRM6. These recombinants delimited the Vg1 functional site to a region of about 7.4 kb (Fig. 4-C). In the B73 reference genome, InDelLM and CRM5 are found at respectively the first intron and the 25th exon of a putative cyclopropane fatty-acid synthase gene (ZmCPA-FAS1,Zm00001d032261) (Fig. 4-D). We thus assigned ZmCPA-FAS as a candidate gene for Vg1.
3.5. Determination of functional site of Vg1 mutation
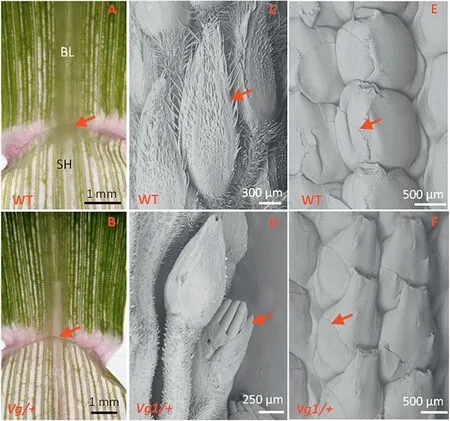
Fig.2-Investigation of the stages at which degenerate ligules and glumes are visible in Vg1 plants.The vestigial ligules begin at the first leaf(A and B),and the reduced glumes of Vg/+plants emerge when the immature tassels are approximately 55 mm long(C and D),and the ears about 51 mm long(E and F).The ligules(A and B)and glumes(C-F)are indicated by arrows.Solid black lines in A and B, and white lines in C-F are scale bars.BL,blade;SH,sheath.
The ZmCPA-FAS1 gene spanned an approximately 13-kb region consisting of 25 exons and 24 introns (Fig. 4-D). To identify the mutation site, several gene-specific primers(Table S4) were designed to amplify ZmCPA-FAS1 from the Vg1 mutant. When all the DNA fragments covering ZmCPAFAS1 were cloned except the sixth intron, TAIL-PCR was employed in combination with a Vg1 genomic fosmid library to ascertain the sequence information in the sixth intron.Finally, an 802-bp fragment containing the first half of the sixth intron of ZmCPA-FAS1 and an additional sequence were obtained by TAIL-PCR (Fig. S3), and a 3949-bp fragment consisting of an additional sequence and the second half of the sixth intron was obtained by subcloning a positive fosmid clone(Fig.S4).The additional sequences were found to insert between the bases A and T.The presence of a conserved CTAG sequence at the 3′end of the insertion suggested that it was a Helitron transposon [17]. The identification of the insertion as a Helitron transposon was further confirmed by a BLAST search of the insertion sequence against the maize transposable element database, which showed that the 5′- and 3′-terminal ends had high similarity with the Helitron subfamily Hip1 (Fig. S5). The DNA fragments corresponding to the insertion site in Vg1 in 71 inbred maize lines were sequenced(Fig.S6), and no foreign insertions were detected.Besides the Helitron insertion in Vg1, other genetic variation was also investigated in 7.4-kb DNA fragment of ZmCPA-FAS1,but most variation was also present in its alleles in Mo17(Zm00014a001048), PH207 (Zm00008a003822), W22(Zm00004b003724), F7 (Zm00011a003544), EPI(Zm00010a003577), and CML247 (GRMZM2G079805). We accordingly speculated that the Helitron insertion in ZmCPAFAS1 caused the mutant phenotype.
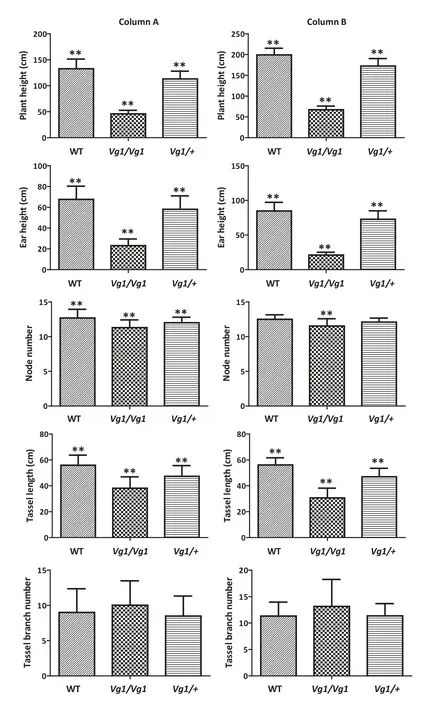
Fig.3-Agronomic trait evaluation in the BC3F2 and BC3F3 populations.(Column A)A total of 42,30,and 45 individuals from the BC3F2 population were evaluated for each genotype of the WT,Vg1/Vg1,and Vg1/+plants,respectively.(Column B)Thirty-six plants of each genotype were evaluated in the BC3F3 population.The rectangles filled with wavy lines,black dots and straight lines represent the trait values of WT,Vg1/Vg1,and Vg1/+ plants,respectively.The double asterisk indicates that the trait values differ highly significantly from the others at the 0.01 probability level.
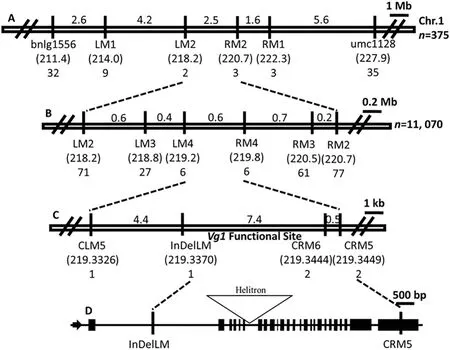
Fig.4- Schematic diagram for map-based cloning of Vg1.(A)A total of 375 BC3 plants were used for screening for recombinants.Vg1 was initially mapped at a chromosome region of about 2.5 Mb flanked by LM2 and RM2 in bin 1.07 on the long arm of chromosome 1.The numbers above the lines and in parentheses indicate respectively the physical distances between adjacent markers and the physical positions(Mb)of the markers.The bottom numbers indicate the numbers of recombinants identified at the specific loci.(B)A total of 11,070 plants were used for identifying plants with crossovers.Using the newly developed SSR markers,the Vg1 functional site was delimited to an interval of approximately 0.6 Mb flanked by LM4 and RM4.(C)Using three CAPS markers and one InDel marker,the Vg1 functional site was finally delimited to a region of approximately 7.4 kb, flanked by InDelLM and CRM6.(D)The gene structure of Zm00001d032261 and the exact positions of InDelLM and CRM5 in the gene.
3.6. Helitron insertion leads to misexpression of the ZmCPAFAS1 gene in Vg1 plants
To obtain the full-length cDNA sequences of ZmCPA-FAS1 in B73 and homogenous Vg1 plants,rapid amplification of cDNA ends was performed.Compared with the predicted transcripts of Zm00001d032261 in Gramene, the pre-mRNA was found to be abnormally alternatively spliced in the Vg1 mutant(Fig.5).The second, third, eighth, and ninth introns in Vg1 that were supposed to be spliced from the pre-mRNA were retained as exons in a few transcripts.Vg1 transcripts in the Mo17 genetic background were also investigated, revealing a similar phenomenon (data not shown) and further confirming that the Helitron insertion was responsible for Vg1 misexpression and excluding an effect of genetic background on the gene transcription.
For further investigation of the expression level of ZmCPAFAS1 in Vg1/+ and wild-type plants, total RNA was extracted from their roots, stems, leaves, immature tassels (1-2 cm),immature ears (1-2 cm), and kernels at 20 days after pollination.The primer QP was designed for qRT-PCR,and primers T-1 and T-2 for semi-quantitative RT-PCR (Table S4). The expression pattern indicated that ZmCPA-FAS1 was expressed in all maize tissues but the kernel (Fig. 6). ZmCPA-FAS1 was dominantly expressed in immature tassels and ears, highly expressed in leaves,and weakly expressed in roots and stems.A dramatic change in the gene expression pattern in Vg1/+plants was observed. Expression was reduced in immature ears,stems,and roots and significantly upregulated in leaves.However, no change was observed in immature ears. Semiquantitative RT-PCR indicated that some alternatively spliced forms were expressed exclusively in immature tassels and ears(Fig.S7).The expression of the alternatively spliced forms detected by T-1 was markedly downregulated in Vg1 plants,whereas some alternatively spliced forms detected by T-2 were increased in Vg1 plants. The combined results of qRTPCR and semi-quantitative RT-PCR show clearly that ZmCPAFAS1 was misexpressed in Vg1.
4. Discussion
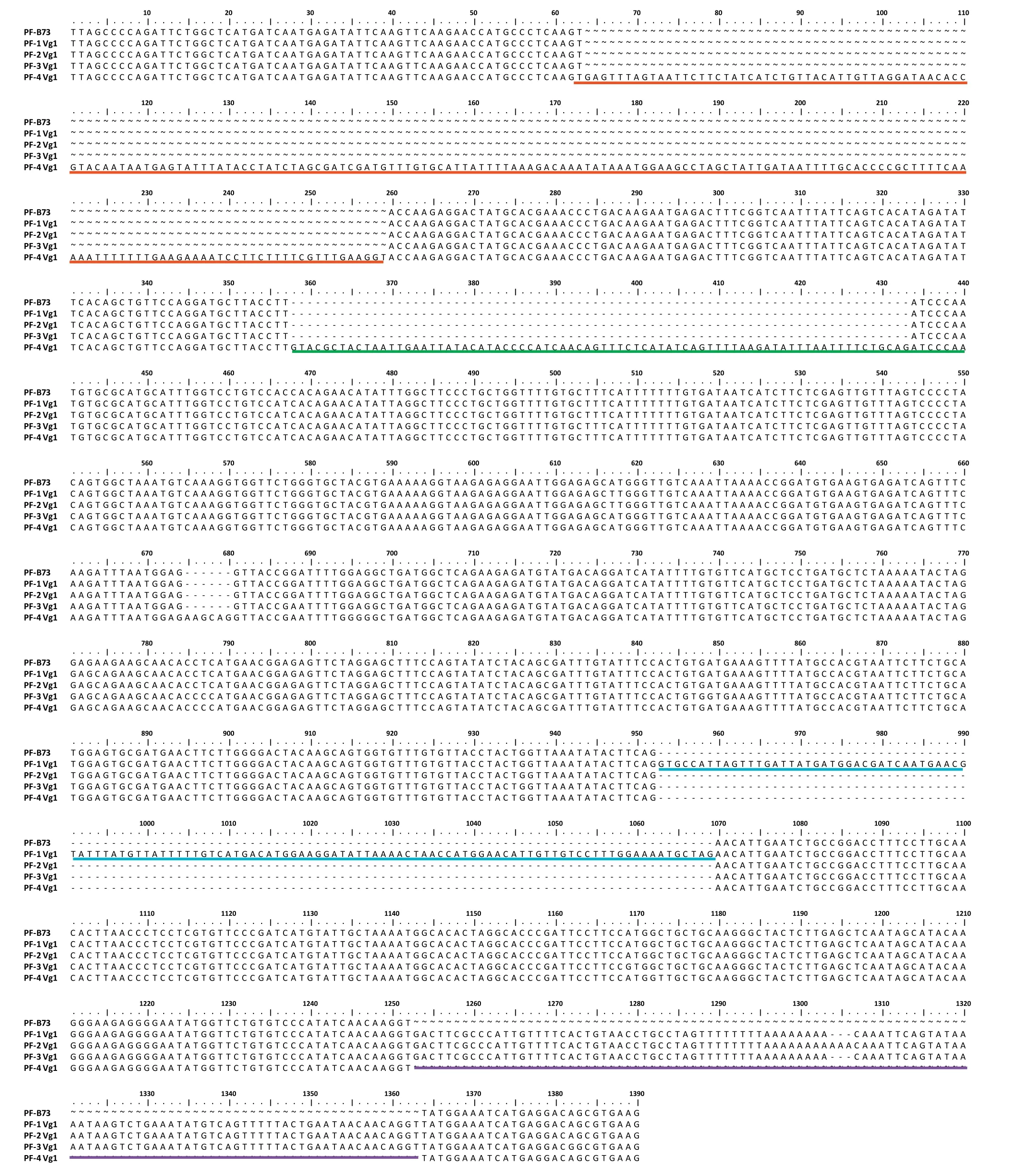
Fig.5-Abnormal transcripts of ZmCPA-FAS1 produced in the homozygous Vg1 mutant.The transcripts are partial fragments of ZmCPA-FAS1 obtained by 5′-and 3′-RACE.The sequences underlined in red,green,blue,and purple represent respectively the second,third,eighth,and ninth introns of ZmCPA-FAS1,which survived the process of alternative splicing.
The maize glume is an important organ that has been subjected to strong artificial selection during maize domestication. Maize morphology has changed drastically from the wild progenitor of maize to its modern cultivated varieties.Determining the functions of the tga1 and Tu1 genes enabled a broader understanding of glume development and maize domestication [4,8]. The glumeless Vg1 mutant was once considered to be a dominant mutant. The greatly reduced glume size results in a lack of pollen shedding and in exposed kernels that are easily shattered.Occasionally,Vg1 plants can produce pollen. In the offspring of self-pollinated Vg1/+plants, the plant height, ear height, and tassel length were significantly different among the different genotypes of Vg1/Vg1, Vg1/+, and vg1/vg1.Vg1 is thus a semi-dominant gene with pleiotropic effects. From an evolutionary perspective,Vg1 plants would not survive in the natural environment,and Vg1 has not conferred beneficial agronomic traits on maize for hybrid seed production in maize breeding history. Furthermore, the domestication locus tga1 is not differentially expressed in the Vg1 mutant (Fig. S8), suggesting that these genes do not function in the same pathway. It can thus be inferred that Vg1 is not a domestication locus strongly selected by either natural or artificial selection.
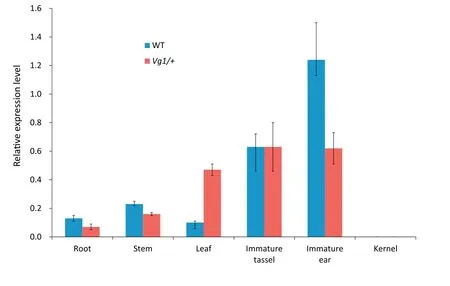
Fig.6-Expression level of ZmCPA-FAS1 in the WT and Vg1/+plants.The expression pattern of ZmCPA-FAS1 is clearly changed in Vg1/+plants.
Our fine-mapping results suggested that ZmCPA-FAS1 is the gene underlying the mutant phenotype. Comparison of genomic DNA sequences revealed a Helitron insertion in the sixth intron of ZmCPA-FAS1. Helitrons are a class of transposable elements that are abundant in maize [18] and often capture and mobilize gene fragments across the maize genome [19,20]. Helitrons potentially play an important role in the synthesis of new genes and the evolution of new proteins [21]. The gene fragments trapped by Helitrons can be transcribed,forming chimeric transcripts consisting of multigene coding regions. Subsequently, these transcripts are sometimes subjected to alternative splicing to produce different transcript variants through differential utilization of splice sites [20,22]. Helitron distribution in maize shows severe biases, and they are usually not inserted into genes[23]. However, in the Vg1 mutant, a member of the Hip1 subfamily, inserted into ZmCPA-FAS1, causes the target gene to be misspliced and misexpressed (Fig. 5 and Fig. 6). Thus,Vg1 is valuable for studying Helitron characteristics and their effects on gene expression.
CPA-FAS catalyzes the biosynthesis of cyclopropane fatty acids (CPA-FAs) by transferring a methyl group from Sadenosylmethionine to a double bond of unsaturated fatty acids(UFAs)in bacteria and plants[24-26].It helps to reduce cell membrane fluidity [27] and improve the ability of bacteria to adapt to different stresses [28-30]. Loss of function of CPA-FAS greatly reduced the survival rate of bacteria exposed to adverse conditions [31]. In plants, the functions of CPA-FAS have been studied [25,32,33]. Unlike in bacteria, after CPA-FAS-mediated catalysis of UFAs, the products were further used as substrates to produce cyclopropenoid fatty acids (CPE-FAs) [34], which are major components of seed oils [35]. Seed oils, especially cotton oil,are extensively consumed by humans worldwide. The consumption of edible oils with abundant CPE-FAs can cause the accumulation of hard fats and other physiological disorders[36,37].Thus,the CPA-FAS gene is a desirable target for genetic modification to eliminate CPE-FAs from oils,thereby improving their quality for human consumption.However, to our knowledge, there has been no previous research on the regulation of maize development by the CPAFAS gene,especially with respect to glume development.Vg1 was previously employed in hybrid sweet corn production for its glumeless character, which made the kernel more easily bitten off the cob [38,39]. However, its presence was also accompanied by adverse traits, such as male sterility and tassel length decrease. Modifying the ZmCPA-FAS1 gene to reduce ear glume size without causing these other negative effects is critical to improving maize production.
Our study will be useful for future research aimed at deciphering the functional mechanisms of Vg1 regulation of ligule and glume development, and further using the glumeless character to breed maize varieties with reduced glume size for improved commercial production of sweet corn.
Supplementary data
Supplementary data for this article can be found online at https://doi.org/10.1016/j.cj.2019.04.001.
Acknowledgments
This work was supported by the Commercial Breeding Innovation of Sweet Glutinous Maize Varieties(cstc2016shms-ztzx80014), Fundamental Research Funds for the Central Universities (XDJK2018C052), and the Major Project Research of Chongqing (cstc2016shms-ztzx80016,cstc2016shms-ztzx80017).

- The Crop Journal的其它文章
- OstMAPKKK5,a truncated mitogen-activated protein kinase kinase kinase 5,positively regulates plant height and yield in rice
- Mapping QTL affecting the vertical distribution and seed set of soybean[Glycine max(L.) Merr.]pods
- Identifying key traits in high-yielding rice cultivars for adaptability to both temperate and tropical environments
- Deep genotyping of the gene GmSNAP facilitates pyramiding resistance to cyst nematode in soybean
- Draft genome sequence of a less-known wild Vigna: Beach pea (V. marina cv. ANBp-14-03)
- Primary metabolite contents are correlated with seed protein and oil traits in near-isogenic lines of soybean