
Expression of long non-coding RNAs in complete transection spinal cord injury: a transcriptomic analysis
2020-01-18 06:02:48LuDingWenJinFuHongYanDiXiaoMinZhangYuTianLeiKangZhenChenTaoWangHongFuWu
中國神經(jīng)再生研究(英文版) 2020年8期
Lu Ding , Wen-Jin Fu , Hong-Yan Di , Xiao-Min Zhang Yu-Tian Lei, Kang-Zhen Chen Tao Wang, Hong-Fu Wu
1 Institute of Stem Cells and Regenerative Medicine, Department of Physiology, Guangdong Medical University, Dongguan, Guangdong Province, China
2 Scientific Research Center, the Seventh Affiliated Hospital of Sun Yat-Sen University, Shenzhen, Guangdong Province, China
3 Clinical Laboratory, Houjie Hospital of Guangdong Medical University, Dongguan, Guangdong Province, China
4 Department of Neurology, Dalingshan Hospital, Dongguan, Guangdong Province, China
5 Department of Hand & Foot Surgery, Houjie Hospital of Guangdong Medical University, Dongguan, Guangdong Province, China
6 Department of Surgery, the Third Hospital of Guangdong Medical University (Longjiang Hospital of Shunde District), Foshan, Guangdong Province, China
Abstract Long non-coding RNAs (lncRNAs) are abundantly expressed in the central nervous system and exert a critical role in gene regulation via multiple biological processes. To uncover the functional significance and molecular mechanisms of lncRNAs in spinal cord injury (SCI), the expression signatures of lncRNAs were profiled using RNA sequencing (RNA-seq) technology in a Sprague-Dawley rat model of the 10th thoracic vertebra complete transection SCI. Results showed that 116 of 14,802 detected lncRNAs were differentially expressed, among which 16—including eight up-regulated (H19, Vof16, Hmox2-ps1, LOC100910973, Ybx1-ps3, Nnat, Gcgr, LOC680254) and eight down-regulated (Rmrp, Terc, Ngrn, Ppp2r2b, Cox6a2, Rpl37a-ps1, LOC360231, Rpph1)—demonstrated fold changes > 2 in response to transection SCI. A subset of these RNA-seq results was validated by quantitative real-time PCR. The levels of 821 mRNAs were also significantly altered post-SCI; 592 mRNAs were up-regulated and 229 mRNAs were down-regulated by more than 2-fold. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses showed that differentially expressed mRNAs were related to GO biological processes and molecular functions such as injury and inflammation response, wound repair, and apoptosis, and were significantly enriched in 15 KEGG pathways, including cell phagocytosis, tumor necrosis factor alpha pathway, and leukocyte migration. Our results reveal the expression profiles of lncRNAs and mRNAs in the rat spinal cord of a complete transection model, and these differentially expressed lncRNAs and mRNAs represent potential novel targets for SCI treatment. We suggest that lncRNAs may play an important role in the early immuno-inflammatory response after spinal cord injury. This study was approved by the Administration Committee of Experimental Animals, Guangdong Province, China.
Key Words: cell apotosis; complete transection injury; high throughput sequencing; inflammation; ischemia related factor vof-16; long non-coding RNA; secondary damage; spinal cord; TNF signaling; transcriptomes
Introduction
Traumatic spinal cord injury (SCI) is a life-changing event with an extremely poor prognosis. It results in physiological impairment and multisystem malfunction including disabilities, intractable neuropathic pain, and extensive potential complications—and thus presents great challenges for patients, carers, and clinicians (Assinck et al., 2017; Kim et al., 2017; Li et al., 2019). To date, no effective treatments for SCI are available because it involves highly complicated pathophysiologic processes and the joint actions of multiple mechanisms. An improved knowledge of SCI pathogenesis would be of great significance for the development of effective therapies and the improvement of curative effects.
Widespread aberrant gene expression occurs during the development and pathological processes of the central nervous system (CNS) (Chandran et al., 2017). Decoding the genomic language that governs the architecture and function of the CNS presents great promise for helping to understand the pathogenesis and therapies of diseases (Ponomarev et al., 2013; Harries, 2019). Long non-coding RNAs (lncRNAs) are the most numerous, heterogeneous, and biologically complex class of non-coding RNAs in the mammalian transcriptome (Hart and Goff, 2016). Earlier studies reported that the majority of lncRNAs were nonfunctional illegitimate transcripts that arose from stochastic/promiscuous promoters. However, increasing evidence has now demonstrated that lncRNAs have a prominent role in essential processes including DNA replication, chromatin shaping, transcription, posttranslational modification of proteins (Hart and Goff, 2016; Chandran et al., 2017; Yang et al., 2019). lncRNAs are also expressed in a highly cell- and tissue-specific manner. Of note, a large proportion of lncRNAs are preferentially expressed within the nervous system, and are associated with distinct neuroanatomical loci (Qureshi and Mehler, 2013; Roberts et al., 2014), suggesting they are intimately linked to the development and pathogenesis of neurological disorders. Many studies have illustrated wide-ranging alteration of lncRNAs in several CNS diseases (Ding et al., 2016; Liu et al., 2017; Quan et al., 2017; Zhou et al., 2017; Wang et al., 2019), and our knowledge of lncRNAs has grown exponentially within the last decade. Nevertheless, most lncRNAs remain to be annotated, and their biological functions and significance have yet to be fully elucidated.
To identify novel targets for further investigation of SCI, we profiled the expression of lncRNAs and mRNAs during spinal cord response to transection injury using whole transcriptome sequencing and subsequent bioinformatics analysis. We validated these results by quantitative real-time polymerase chain reaction (qRT-PCR).
Materials and Methods
Animals
Adult specific pathogen-free Sprague-Dawley (SD) rats (half male/half female), aged 8-10 weeks and weighing 180-200 g, were purchased from Nanfang Medical University, China (license No. SCXK (Yue) 2016-0041). Rats were housed in standard cages under a regulated environment (12-hour light/dark cycle) with free access to food and water. All procedures using laboratory animals were conducted in compliance with the Guide for the Care and Use of Laboratory Animals from the National Institutes of Health and approved by the Administration Committee of Experimental Animals, Guangdong Province, China.
Rat model of transection SCI
Animals were randomly divided into two groups: normal (n = 12) and SCI (n = 12; 7 days post-injury). Transection SCI was conducted as previously reported (Wu et al., 2013). In brief, rats were anesthetized with isoflurane (Coulbourn, Holliston, MA, USA) and fixed in a prone position. The back hair was shaved and a 2- to 3-cm incision was made at the mid-line over the spinous processes from the 9ththoracic spinal vertebra (T9) to T11. The paravertebral muscles were then separated from the vertebrae to expose the T10 vertebra. Subsequently, the T10 spinous process and laminar were surgically removed by laminectomy, and the spine was exposed without disrupting the dura. After stabilizing the spine with the spinous processes of T9-11, the exposed T10 was completely transected using a sharp scalpel to produce a reliable transection SCI model. The muscles and skin were closed in layers post-hemostasis. During surgery, rats were placed on a heating pad to maintain body temperature at 37°C. After revival from anesthesia, rats were kept under specific pathogen-free conditions. Animals were intraperitoneally injected with Penicillin-Streptomycin (10,000 U/mL) daily and given manual bladder evacuations twice a day post-surgery. Rats in the normal group were housed in the same regulated environment (12-hour light/dark cycle) with free access to food and water.
Rats in the normal group and SCI group (7 days post-SCI) were sacrificed at 7 days, and a 5-mm of spinal cord containing the injury epicenter from SCI rats or corresponding spinal segment in normal rats were isolated quickly for RNA sequencing (RNA-seq) (n = 6/group) and qRT-PCR (n = 6/group).
Total RNA extraction
Total RNA from T10 spinal cord segments was extracted by Trizol reagent (Invitrogen, Carlsbad, CA, USA) in accordance with the manufacturer’s protocol. DNA contained in tissues was removed using the DNase I Mini Kit (Qiagen, Hilden, Germany). RNA quantity was determined spectrophotometrically at optical density (OD)260and OD260/OD280= 1.8-2.1 using a NanoDrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA).
RNA sequencing
RNA samples collected from normal rats and damaged spinal cords at 7 days post-transection SCI were used for sequencing analysis. Transcriptome sequencing of RNA harvested from normal rats and SCI animals was performed by Illumina HiSeq 2500 (RIBO Biotech Company, Guangzhou, China). Briefly, ribosomal RNAs were removed, and purified mRNAs were fragmented into short fragments. Random hexamer-primers were used to synthesize complementary DNAs from these short mRNA fragment templates. After end-repair and ligation of Illumina sequencing adaptors, size-suitable fragments were selected and purified for qRTPCR amplification with SYBR Green Kit (Takara, Tokyo, Japan) on Illumina EcoTM(Illumina, San Diego, CA, USA). The amplified library was then sequenced using Illumina HiSeq 2500 (Additional file 1).
Primary sequencing data (raw reads) were subjected to quality control to filter out low-quality reads, including reads that contained > 10% N-base calls, and low-quality reads accounted for more than 20% of the whole transcriptome. The expression levels of mapped genes were calculated by the reads per kilobase transcriptome per million mapped reads method to normalize gene expression levels. Transcripts that had a fold change > 2 and q-value < 0.05 were considered to be significantly differentially expressed.
Bioinformatics analysis
The differentially expressed transcripts were entered into the Database for Annotation, Visualization and Integrated Discovery (DAVID; http://david.abcc.ncifcrf.gov/). Gene Ontology (GO) annotation was used to identify the molecular functions of the genes. GO encompasses three domains: biological process, cellular component and molecular function, and provides extensive annotation of genes and gene products (http://www.geneontology.org). Pathway analysis was also used to identify the potential functions of differentially expressed mRNAs according to the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (http://www.genome.jp/kegg/). The P-values denote the significance of GO term enrichment or the significance of the KEGG pathway correlation (P-value < 0.05 was considered to be statistically significant).
Quantitative real-time PCR assay
LncRNAs from the RNA-seq analysis were selected for validation by qRT-PCR. Total RNA was reverse-transcribed using SuperScriptTMIII reverse transcriptase (Invitrogen) according to the manufacturer’s protocol, with random primers and 1 μg RNA from the same samples used for RNASeq. qRT-PCR was conducted using the SYBR-Green q-PCR MasterMix (Arraystar, Rockville, MD, USA) in a ViiA 7 Real-time PCR System (Applied Biosystems, Waltham, MA, USA). The oligonucleotide sequences of the lncRNA primers are in Table 1. Each qRT-PCR reaction included 5 μL Master Mix, 0.5 μL forward primer, 0.5 μL reverse primer, and 2 μL complementary DNA. The total volume was adjusted to 10 μL with double-distilled H2O. The following thermocycler parameters were used to generate the dissociation curve: (1) 95°C for 10 minutes; (2) 40 cycles of 95°C for 10 seconds, 60°C for 60 seconds; and (3) 60°C to 99°C. LncRNA expression was normalized using glyceraldehyde-3-phosphate dehydrogenase. Relative quantification of lncRNAs was calculated by the 2-ΔΔCtmethod. Glyceraldehyde-3-phosphate dehydrogenase was used as an internal control.

Table 1 Oligonucleotide sequences of the quantitative real-time polymerase chain reaction primers
Statistical analysis
Comparison of the normal group and SCI group (7 days post-SCI), in the RNA-seq analysis was evaluated using fold change and Student’s t-test in the IBM SPSS Statistics 25.0 software (IBM, Armonk, NY, USA). A false discovery rate was calculated to correct the P-value. A fold change ≥ 2 and P-value ≤ 0.05 was considered the threshold value to designate differentially expressed lncRNAs and mRNAs.
Results
mRNA expression profiles in SCI
RNA-seq detected differential expression of 12,400 mRNAs between SCI animals and normal animals, of which 9219 mRNAs were up-regulated and 6786 mRNAs were down-regulated. Further analysis indicated 591 significantly up-regulated mRNAs and 229 significantly down-regulated mRNAs post-SCI (P < 0.05, |log2(fold change)| > 1) (Table 2). The heat map indicates the expression profiles of all mRNAs (Figure 1A). The volcano plot shows the mRNAs significantly expressed between the two groups (Figure 1C).
lncRNA expression profiles in SCI
Following hierarchical clustering, 14,802 lncRNAs were identified by high throughput RNA-seq, including 491 annotated lncRNAs and 14,311 novel ones (Table 2). Among the 491 known lncRNAs, the expression levels of 16 (eight up-regulated and eight down-regulated) were greatly altered after SCI compared with the normal group, according to a P-value (< 0.05) and fold change (> 2). A heat map displaying the expression signatures of lncRNAs before and after SCI is illustrated in Figure 1B. The volcano plot shows the significantly differentially expressed lncRNAs (Figure 1D). We observed that differentially down-regulated lncRNAs were more common than up-regulated lncRNAs, whereas this trend was opposite in our mRNA dataset. A heat map and hierarchical cluster analysis of differentially expressed lncRNAs is illustrated in Figure 2. Table 3 lists the seven most-differentially expressed lncRNAs (P < 0.05, |log2(fold change)| > 3) following SCI.
GO and KEGG pathway analysis of differentially expressed mRNAs in SCI rats
GO enrichment analysis was performed to investigate the effect of SCI on biological processes, cellular components, and molecular functions. The results demonstrated that 489 biological processes, 74 cellular components, and 58 molecular functions were enriched in the differentially expressed gene set. The ten most-enriched GO biological processes were mainly associated with responses to damage and inflammation, including response to external stimulus, response to wounding and stress, inflammatory response, and defense response (Figure 3A). Of note, biological processes such as apoptosis, tumor necrosis factor (TNF) production, gliogenesis, regulation of synaptic plasticity, and vasculogenesis were also significantly down-regulated. The ten most-enriched GO cellular components were cell periphery, plasma membrane, cytoplasm, cell fraction, and cytosolic ribosome (Figure 3B), and the ten most-enriched GO molecular functions included protein binding, cytoskeletal protein binding, insulin-like growth factor binding, and enzyme binding (Figure 3C).
KEGG pathway analysis revealed 15 substantially enriched pathways, many of which are relevant to the inflammatory response, including ribosome, phagosome, TNF signaling pathway-Rattus norvegicus (rat) (Figure 4), leukocyte tran-sendothelial migration, bacterial invasion of epithelial cells, and cell adhesion molecules (Figure 5). As illustrated in Figure 4, several molecules involved in TNF signaling were differentially expressed (red box) post-SCI. These results indicate that the immune inflammatory response in the injury microenvironment exerts a critical role in the early pathological progression of SCI.
Quantitative real-time PCR validation of lncRNA expression
To validate the results from high-throughput RNA-seq, four-teen highly up- or down-regulated lncRNAs were selected for qRT-PCR verification. As shown in Figure 6, although the fold change in lncRNA expression between the two datasets differed, there were consistent tendencies between the RNA-seq and qRT-PCR results. We confirmed the expression dynamics of five down-regulated lncRNAs (such as Rmrp and Terc), and nine up-regulated lncRNAs (such as H19, Vof16 and Hmox2-ps1) (Figure 6), suggesting that these lncRNAs are potentially functional in the pathological development of SCI.

Table 2 Overview of RNA sequencing results in rats after spinal cord injury
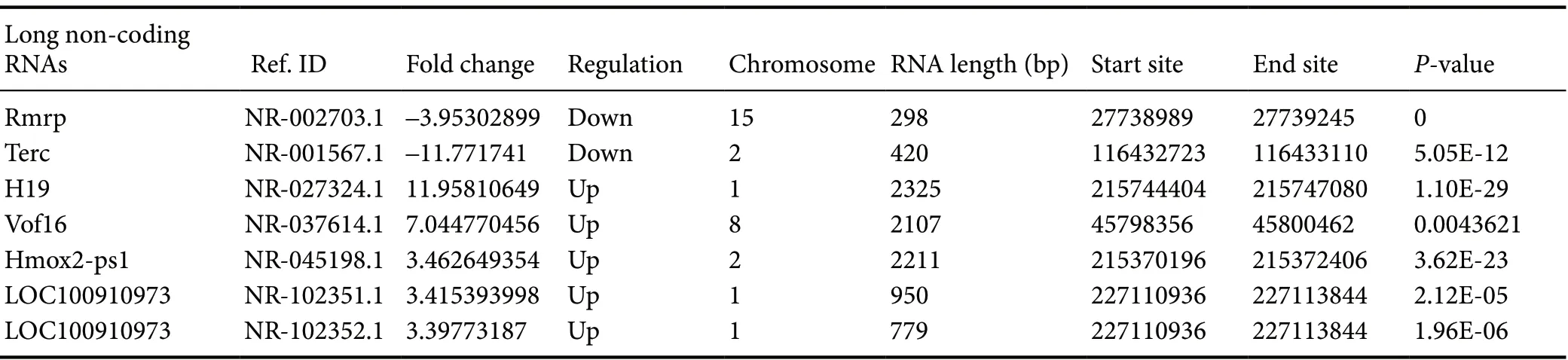
Table 3 Differentially expressed long non-coding RNAs in spinal cord injury rats (|log2(fold change)| > 3)
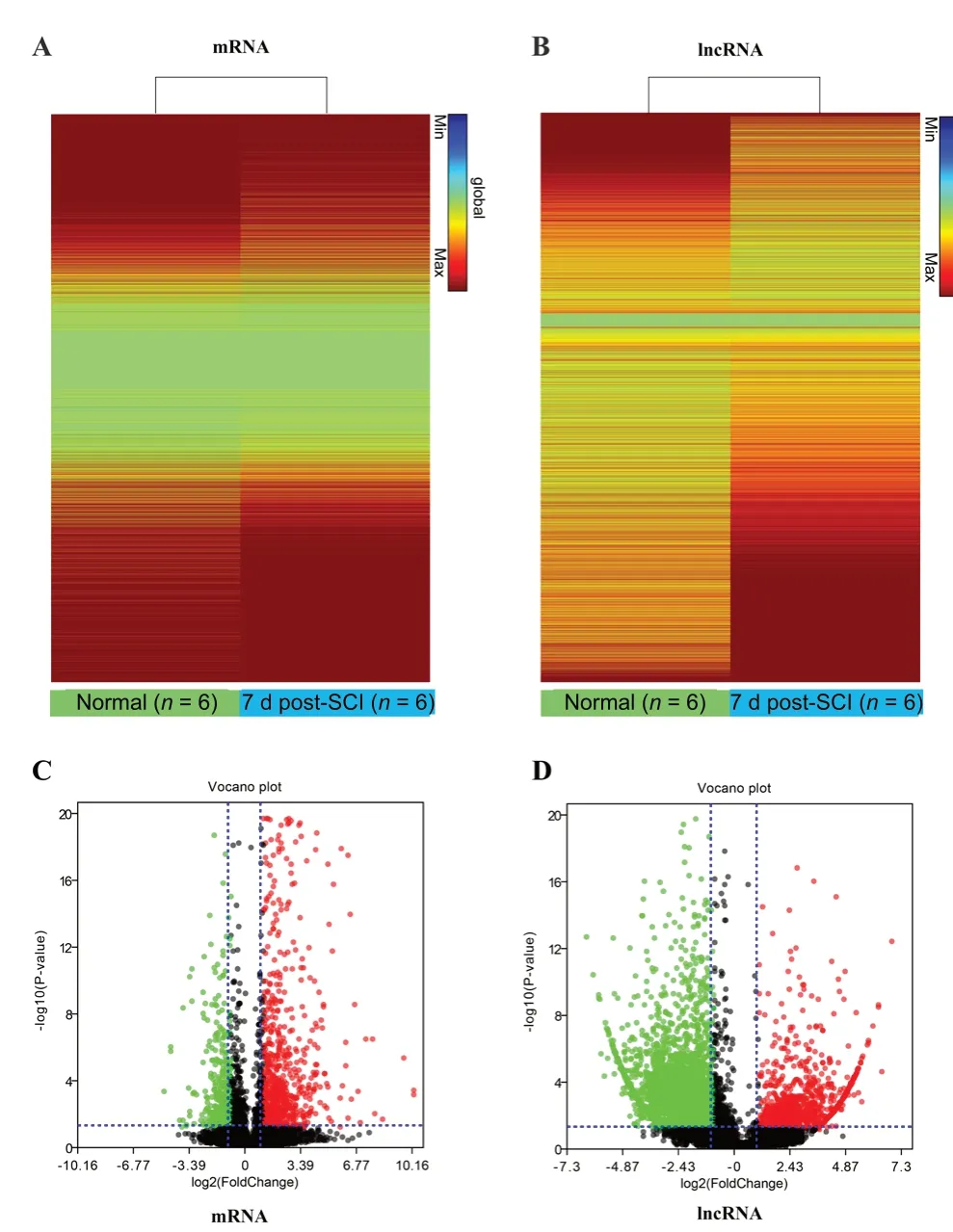
Figure 1 Altered expression profiles of mRNAs and long non-coding RNAs (lncRNA) following transection spinal cord injury (SCI).
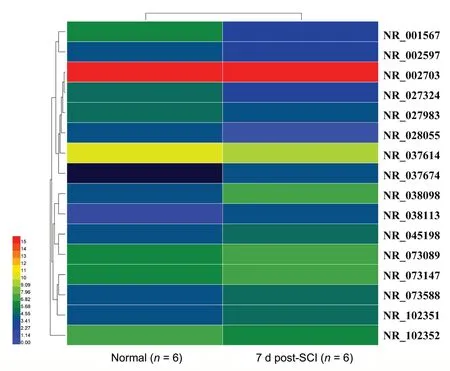
Figure 2 Heat map showing hierarchical clustering analysis of differentially expressed long non-coding RNAs after spinal cord injury (SCI) whose change in expression is more than 2-fold (|log2(fold change)| > 1, P < 0.05).
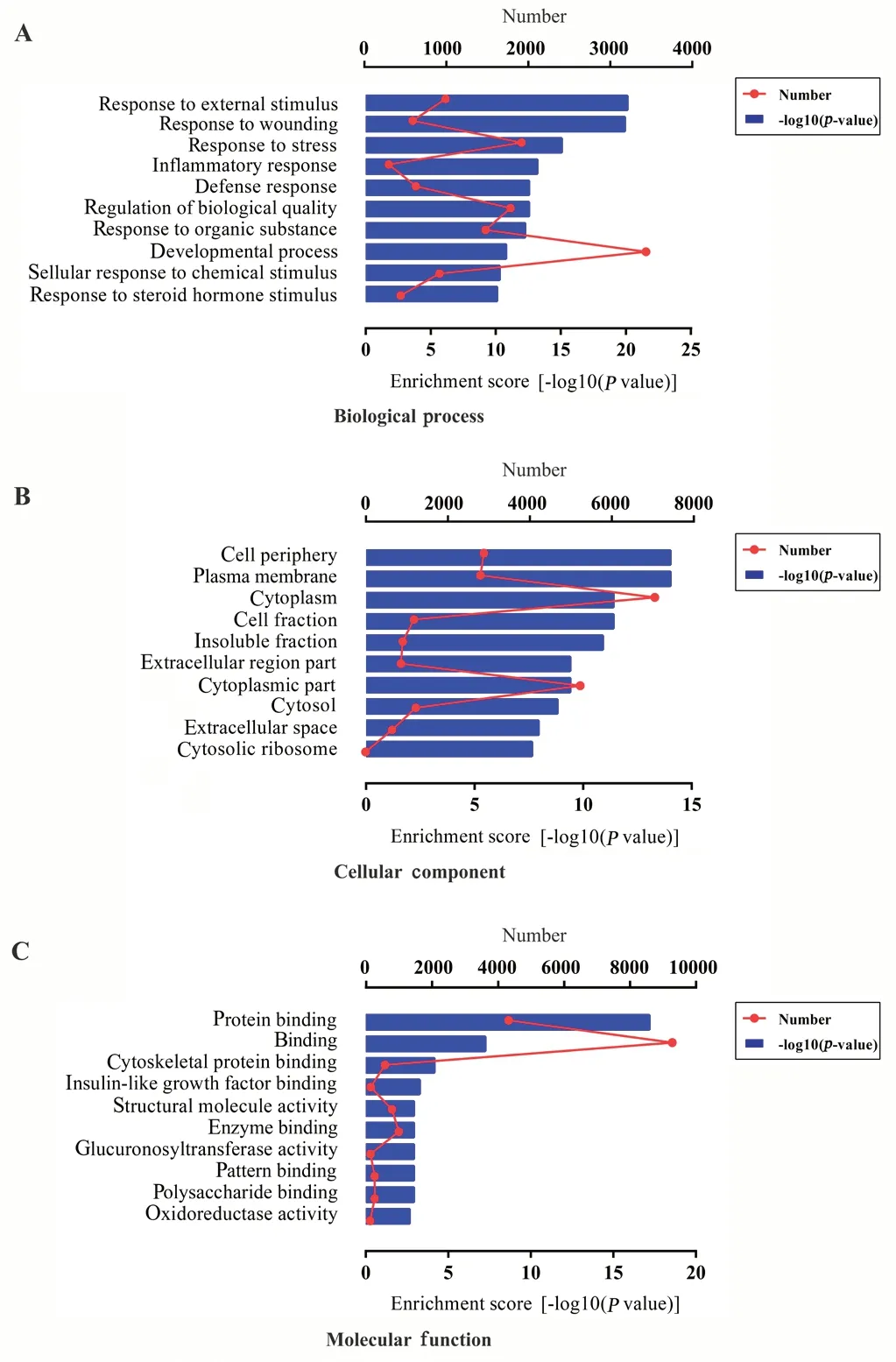
Figure 3 Gene Ontology (GO) enrichment analyses of differentially expressed mRNAs.
Discussion
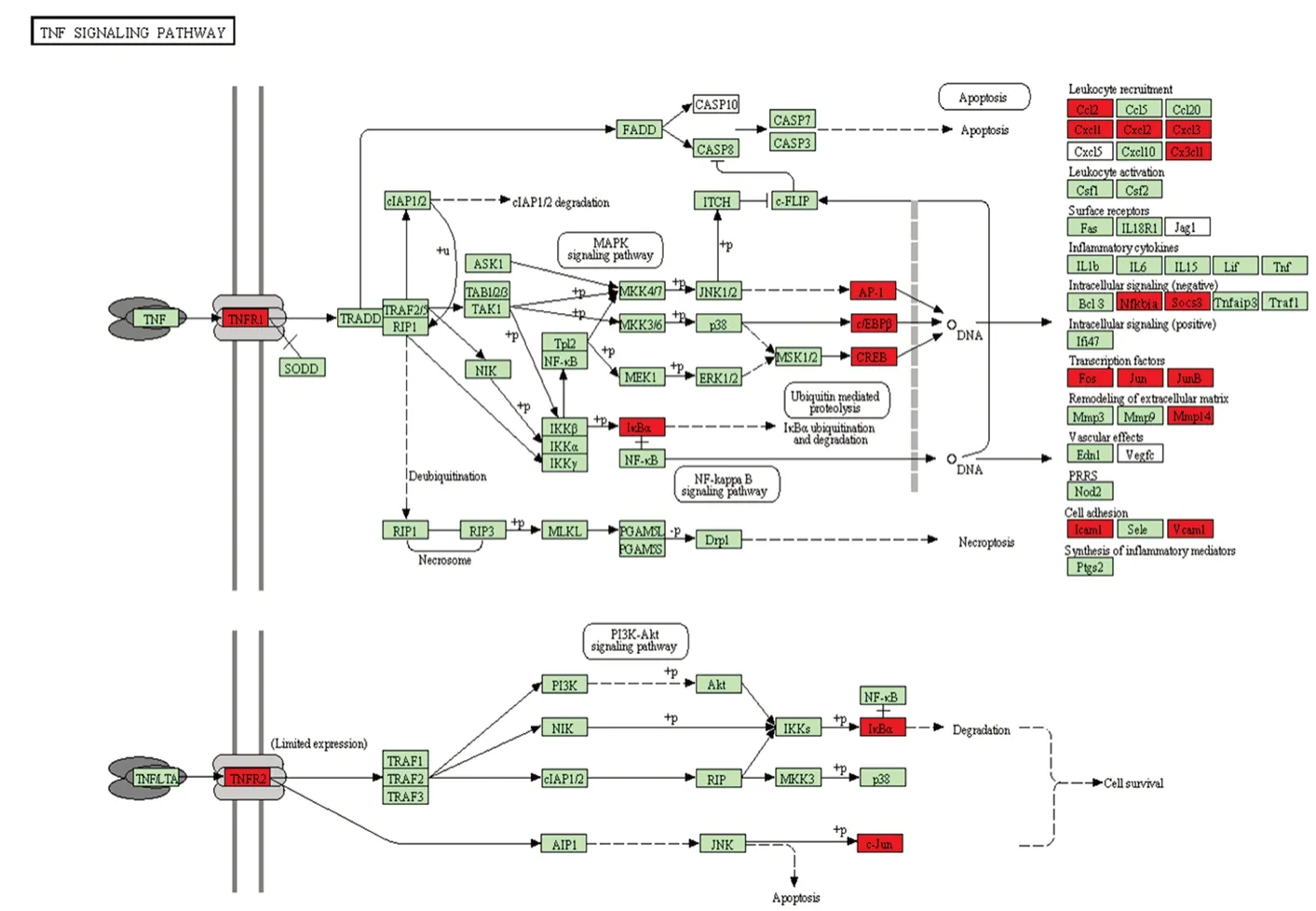
Figure 4 Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis.
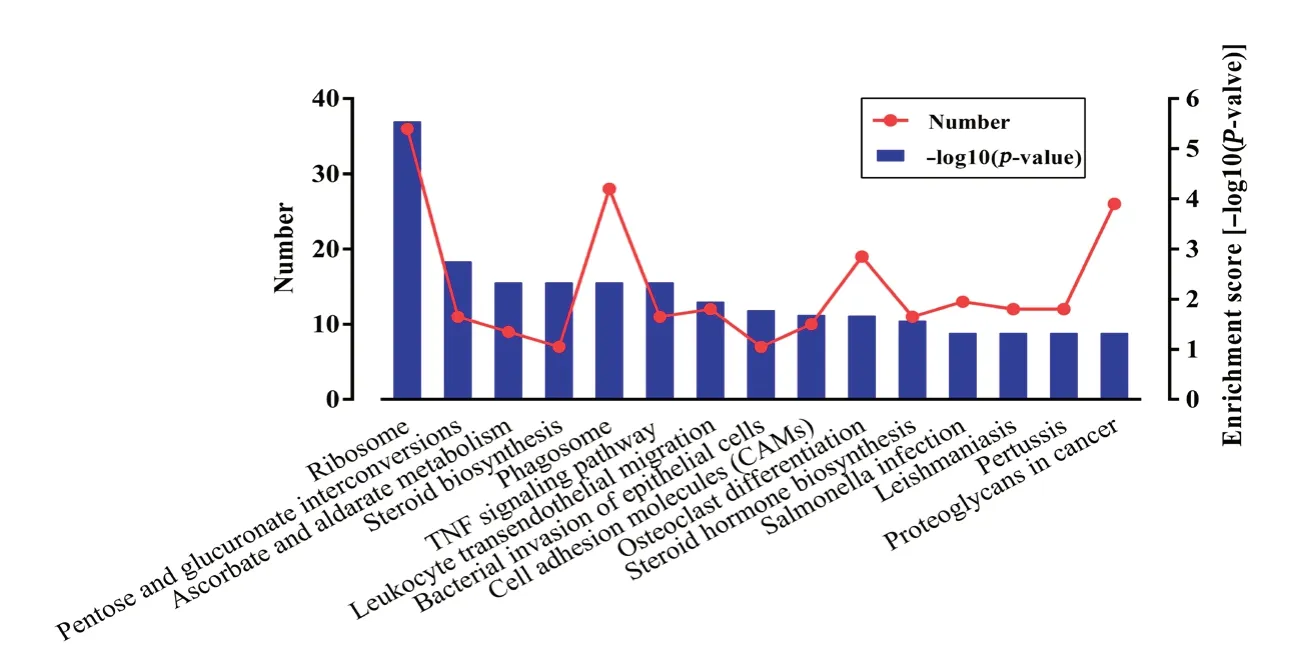
Figure 5 Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of differentially expressed mRNAs.
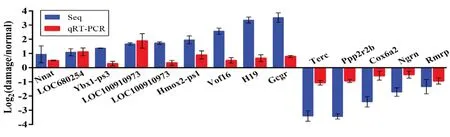
Figure 6 Real-time quantitative polymerase chain reaction (qRT-PCR) validation of 14 differentially expressed long noncoding RNAs (lncRNAs) in the spinal cord of spinal cord injury (SCI) rats.
Traumatic SCI is an incurable condition with limited potential for structural repair and functional recovery. This is due to the lack of therapeutic options available clinically (He and Jin, 2016), and the complex and aggressive pathophysiology of SCI. In recent years, promising effects of lncRNAs in the biological regulation of CNS disease have been widely evidenced. As such, identifying lncRNAs involved in SCI would be of great significance for better understanding its pathogenesis.
A previous study reported a microarray analysis of lncRNAs in contusion SCI; however, the expression profiles of lncRNAs in complete transection SCI remain unknown. Here, we investigated the expression profiles of lncRNAs in SCI rats following T10 transection. As expected, compared with the normal group, an abundance of lncRNAs was either up-regulated or down-regulated post-SCI. Some of these differentially expressed lncRNAs related to specific cellular and signaling events, suggesting that they may be potential pathological markers or therapeutic targets for SCI.
The suitability of the animal model and detection time period exert a determined role in experimental research (Sharif-Alhoseini et al., 2017). In general, it is accepted that contusion SCI is the classic SCI model because of its similarity to the clinical situation—which also comprises bony fragments or extruded disk materials in addition to spinal tissue destruction. However, because of variation in residual tissues, there are shortcomings of the contusion SCI model relating to the evaluation of nerve regeneration and functional recovery. These are not issues in the complete transection SCI model, a standard SCI model with identical severity. Secondary injury following SCI is the core of its debilitating pathologic progression, and provides the optimal time frame for therapeutic interventions. Necrotic and apoptotic cell death in the acute and subacute phase (about 2 weeks post-injury) are the most significant post-trauma changes, and these peak at 7 days after the initial mechanical insult. As research concerning lncRNAs in complete transection SCI has to our knowledge not been reported, we therefore conducted a transection SCI model and analyzed lncRNA expression profiles at 7 days post-injury to get a more extended understanding of lncRNAs pattern in SCI.
In the present study, 14,802 lncRNAs were detected (including 491 annotated lncRNAs) among which eight lncRNAs were up-regulated and eight were down-regulated (P < 0.05, |log2(fold change)| > 2) between normal and SCI animals. To validate these RNA-seq results, qRT-PCR was performed. Twenty-four lncRNAs were randomly selected for qRT-PCR on an independent series of samples taken from both the normal and SCI animals. Of these, 14 exhibited a statistically significant difference that was consistent with that of the high-throughput detection. Specifically, nine lncRNAs, including Nnat, LOC680254, Vof16, and H19, were significantly up-regulated, while five lncRNAs, including rmrp, and Terc, were significantly down-regulated.
Furthermore, GO enrichment and KEGG pathway analyses were used to preliminarily study the potential functions of the differentially expressed mRNAs. In contusion SCI, studies have reported that most altered mRNAs were involved in transport, cell adhesion, metabolic process, and innate immune response; a KEGG analysis revealed neuroactive ligand-receptor interactions, the phosphoinositide 3-kinase-Akt signaling pathway, focal adhesion, and metabolic pathways as the top enrichment pathways (Ding et al., 2016). Conversely, in our GO analysis of transection SCI, many of the differentially expressed transcripts were associated with response to wounding, inflammatory response, immune system process, cell periphery, protein binding, and cytoskeletal protein binding. Similarly, our KEGG enrichment analysis revealed that the transcripts with changed expression were involved in phagosome, TNF signaling pathway, leukocyte transendothelial migration, and cell adhesion molecules—pathways which are mostly associated with immuno-inflammatory responses. This highlights the critical role of the inflammatory reaction cascade in secondary damage following SCI. These results suggest that the different severity of injury may contribute to a difference in the pathological processes in these two types of SCI model rats.
Aggressive inflammation and immune stress response after SCI leads to neuronal death, myelin loss, and microcapsule formation—seriously hindering cell survival and axonal regeneration. TNF is a multifunctional proinflammatory cytokine that plays important roles in various physiological and pathological processes, such as cell proliferation and apoptosis, modulation of immune responses, and induction of inflammation (Song et al., 2018). An increase of TNF-α post-SCI not only results in cell apoptosis and necroptosis via the activation of several signaling pathways, it also activates astrocyte proliferation to accelerate glial scar formation (Esposito and Cuzzocrea, 2011). Accumulating evidence has demonstrated that a blockade of TNF signaling exerts anti-inflammatory action, antinociceptive effects, and neuroprotective properties in many CNS diseases including SCI. In our study, genes involved in the TNF signaling pathway, such as TNF receptor, IkBα, and Jun, were all differentially expressed after SCI. A better understanding of TNF signaling and its relatives may aid in providing new avenues for SCI therapeutic intervention, e.g. by inhibiting and modulating the biological activity of these cytokines.
To date, few lncRNAs have been annotated and functionally characterized. Of these, lncRNA H19 (NR_027324) is abundantly expressed in fetal tissues and adult metabolic organs such as liver, muscle, and adipose , and has been extensively investigated in several genetic disorders and cancers. Notably, recent studies have reported that H19 is also expressed abnormally following hypoxia, where it functions to stimulate acceleration of cerebral ischemia and reper-fusion injury, and is associated with the susceptibility and clinical features of ischemic stroke (Huang et al., 2019). Additionally, increased H19 was detected in Schwann cells in the peripheral neuropathy and it induces hippocampal neuronal apoptosis in diabetes mellitus (Zhao et al., 2017; Han et al., 2018). Similarly, an up-regulation of H19 in our study suggests a possible role in metabolic disturbance and ischemia-induced neuron death after SCI. NR_037614, a lncRNA defined as Rattus norvegicus ischemia related factor vof-16 (Vof16), also drew our attention—despite not having the most significantly changed expression. Vof16 is known to be abundant in the hippocampus, the piriform cortex, and the area around the aorta (Tohda and Watanabe, 2004). Vof16 is also up-regulated in ischemic brain injury, and has been suggested to be related to neuronal damage and cognitive impairment in brain injury, neurodegenerative disease and CNS angiitis; however, to our knowledge, expression enrichment and a potential function of Vof16 in SCI has not been previously reported. We observed significant up-regulation of Vof16, implying it has a possible effect on the neurological impairment that is induced by hypoxic-ischemic damage after SCI, and offers us inspiration for future work.
Consistent with other studies, our bioinformatics analysis identified that the expression of a broad suite of lncRNAs and mRNAs is significantly altered following transection SCI. However, although we have preliminarily predicted lncRNA functions from our sequencing results, it would be premature to use these lncRNAs as possible SCI biomarkers or therapeutic targets. Because of acknowledged limitations of this work, such as the small number of samples and individual differences in RNA-seq data, the biological functions of lncRNAs and their role in the SCI pathological process require further study.
Conclusion
We have demonstrated the expression dynamics of lncRNAs and mRNAs after transection SCI through RNA-seq analysis and qRT-PCR validation. Identification of potential lncRNAs provides new opportunities to study RNA-directed epigenetic regulators and their essential role in SCI pathologies, and to uncover novel targets for lncRNA-based diagnostics and SCI therapeutics in the future.
Author contributions:Study design and funding support: HFW; animal experiment implementation: LD, HYD, YTL; molecular biology experiment implementation: WJF, XMZ, KZC; manuscript writing: LD; manuscript revising: HFW, TW. All authors approved the final version of the paper.
Conflicts of interest:The authors declare that they have no competing interests.
Financial support:This work was financially supported by the National Natural Science Foundation of China, No. 81371366 (to HFW); Characteristic Innovation Project of Colleges and Universities in Guangdong Province of China, No. 2018KTSCX075 (to HFW); the Key Project of Social Development of Dongguan of China, No. 20185071521640 (to HFW); College Students’ Science and Technology Innovation Training Project, China, Nos. 201810571058, GDMU2018024, GDMU2018056, GDMU2018061 (to HFW); College Students’ Innovative Experimental Project in Guangdong Medical University, China, No. ZZDS001 (to HFW); College Students’ Science and Technology Innovation Cultivation Project in Guangdong of China, No. pdjh2019b0217 (to HFW). The funders had no roles in the study design, conduction of experiment, data collection and analysis, decision to publish, or preparation of the manuscript.
Institutional review board statement:All experimental procedures and protocols were approved by the Administration Committee of Experimental Animals, Guangdong Province, China. All experimental procedures described here were in accordance with the National Institutes of Health (NIH) guidelines for the Care and Use of Laboratory Animals (NIH Publication No. 85-23, revised 1996).
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement:Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-Non-Commercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewer:Guillermo García-Alías, Universitat Autonoma de Barcelona, Spain.
Additional file:
Additional file 1:High throughput RNA sequencing (RNA-seq) and genome-wide read mapping.

- 中國神經(jīng)再生研究(英文版)的其它文章
- LIM kinases in synaptic plasticity and their potential as therapeutic targets
- Increased thalamocortical connectivity from the affected thalamus to the unaffected hemisphere in a stroke patient
- Abnormal brain activity in adolescents with Internet addiction who attempt suicide: an assessment using functional magnetic resonance imaging
- Inhibition of BACE1, the β-secretase implicated in Alzheimer’s disease, by a chondroitin sulfate extract from Sardina pilchardus
- Neuroprotective effect of deferoxamine on erastininduced ferroptosis in primary cortical neurons
- Human umbilical cord mesenchymal stem cells to treat spinal cord injury in the early chronic phase: study protocol for a prospective, multicenter, randomized, placebo-controlled, single-blinded clinical trial