
MicroRNA and mRNA profiling of cerebral cortex in a transgenic mouse model of Alzheimer’s disease by RNA sequencing
2021-03-03 15:07:24LiZengHaiLunJiangGhulamMdAshrafZhuoRongLiRuiLiu
中國神經(jīng)再生研究(英文版) 2021年10期
Li Zeng , Hai-Lun Jiang , Ghulam Md Ashraf , Zhuo-Rong Li , Rui Liu
Abstract In a previous study, we found that long non-coding genes in Alzheimer’s disease (AD) are a result of endogenous gene disorders caused by the recruitment of microRNA (miRNA) and mRNA, and that miR-200a-3p and other representative miRNAs can mediate cognitive impairment and thus serve as new biomarkers for AD. In this study, we investigated the abnormal expression of miRNA and mRNA and the pathogenesis of AD at the epigenetic level. To this aim, we performed RNA sequencing and an integrative analysis of the cerebral cortex of the widely used amyloid precursor protein and presenilin-1 double transgenic mouse model of AD. Overall, 129 mRNAs and 68 miRNAs were aberrantly expressed. Among these, eight down-regulated miRNAs and seven up-regulated miRNAs appeared as promising noninvasive biomarkers and therapeutic targets. The main enriched signaling pathways involved mitogen-activated kinase protein, phosphatidylinositol 3-kinase-protein kinase B, mechanistic target of rapamycin kinase, forkhead box O, and autophagy. An miRNA-mRNA network between dysregulated miRNAs and corresponding target genes connected with AD progression was also constructed. These miRNAs and mRNAs are potential biomarkers and therapeutic targets for new treatment strategies, early diagnosis, and prevention of AD. The present results provide a novel perspective on the role of miRNAs and mRNAs in AD. This study was approved by the Experimental Animal Care and Use Committee of Institute of Medicinal Biotechnology of Beijing, China (approval No. IMB-201909-D6) on September 6, 2019.
Key Words: 3′-untranslated region; Alzheimer’s disease; biomarker; cerebral cortex; Gene Ontology; high-throughput sequencing; intracellular neurofibrillary tangles; microtubule-associated protein-τ; miRNA-mRNA network; presenilin 1 Chinese Library Classification No. R446.1; R741.04; Q344+.13
Introduction
Alzheimer’s disease (AD) is the leading cause of dementia, and characterized by cognitive impairment and memory loss (Alexiou et al., 2019; Fiorini et al., 2020; Li et al., 2020). The pathological AD hallmarks are the presence of intracellular neurofibrillary tangles, extracellular senile plaques, and neuronal loss (Xu et al., 2019; Mamun et al., 2020). Patients with familial AD caused by the mutation of specific genes, such as amyloid-beta peptide precursor protein (APP), presenilin 1 (PS1), presenilin 2 (PS2), and microtubule-associated protein-τ (MAPT), are less than 5% (Cacace et al., 2016). However, the morbidity of sporadic AD, characterized by the involvement of multiple molecular mechanisms, is largely enigmatic and greater than 95% (Dorszewska et al., 2016). The drugs currently available to treat AD do not reduce neuronal deterioration or death. Most of the candidate drugs targeting amyloid-beta peptides (Aβ),tau tangles, neurotransmitters, and against neuroinflammation have proved unsuccessful in clinical trials (Bae et al., 2019;Cummings et al., 2019). Consequently, the identification of reliable biomarkers that may contribute to early diagnosis andtimely therapeutic intervention is urgently needed.
MicroRNAs (miRNAs) are single-stranded non-coding RNA of 19-25 nucleotides in length. The biogenesis of miRNAs can be canonical or non-canonical, which involve the endoribonuclease Dicer and the Argonaute protein family,respectively. Both pathways ultimately lead to a functional miRISC complex binding with the target mRNAs to inhibit their translation (Matsuyama and Suzuki, 2019; Xiao and MacRae, 2019). Gene expression is controlled by binding the 3′-untranslated region (UTR) with bases in the mRNAs;this reduces transcription efficiency and/or decreases mRNA expression, which in turn results in a protein production decrease of more than 80% (Guo et al., 2010). Many miRNAs,including miR-346, miR-101, miR-153, miR-15b, miR-339-5p,and miR-200a-3p, contribute to Aβ production and clearance,tau phosphorylation, synaptic dysfunction, and autophagy by suppressing the translation or inducing the degradation of their target mRNAs involved in AD pathogenesis (Martinez and Peplow, 2019; Wang et al., 2019a; Hou et al., 2020; Rodriguez-Ortiz et al., 2020). However, the physiological and pathological function of the aberrantly expressed miRNAs and mRNAs involved in AD is not yet sufficiently clear.
In this study, RNA sequencing was achieved to identify the profile of miRNA and mRNA expression involved in AD progression in APP/PS1 double transgenic mice of different ages compared with age-matched wild type (WT) control mice.Subsequently, the potential target genes of the significantly dysregulated miRNAs, their biological function, and pathway enrichment were assessed. Furthermore, the miRNA-mRNA network was constructed to identify novel diagnostic AD biomarkers and therapeutic targets.
Materials and Methods
Animal treatment and sample preparation
All experiments were designed and reported according to the Animal Research: Reporting ofIn VivoExperiments (ARRIVE)guidelines. The APP/PS1 transgenic mice and age-matched WT littermates were purchased from the Zhishan Healthcare Research Institute (Beijing, China; license No. SCXK2019-0008). The Experimental Animal Care and Use Committee of Institute of Medicinal Biotechnology of Beijing, China (approval No. IMB-201909-D6) approved the animal experiments(approval No. IMB-201909-D6) on September 6, 2019. Twelve APP/PS1 mice were grouped by age (1 month, 3 months,6 months, and 9 months), and the same grouping was applied to the twelve corresponding WT control mice. Each age group included three mice (two female and one male).Before performing the RNA sequencing, APP/PS1 mice were subjected to the Morris water maze test (Liu et al., 2018), in which their learning and memory dysfunction was evaluated using a water navigation task and exploration of the space,and compared with that of the WT control mice (Additional Figure 1). Mice were then sacrificed by cervical dislocation and the cerebral cortex was stored in liquid nitrogen.
RNA extraction
We isolated the total RNA from the cerebral cortices using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. The total RNA concentration was assessed using a Spark 20M multimode microplate reader(Tecan Group Ltd., Mannedorf, Switzerland). The integrity of RNA was evaluated using 1% agarose gel electrophoresis.The RNA concentration and electrophoresis are shown inAdditional Table 1andAdditional Figure 2.
mRNA library construction and sequencing
One microgram of total RNA was used for complementary DNA (cDNA) library preparation. Subsequently, 150-200 bp cDNA fragments were enriched and purified using the AMPure XP system (Beckman Coulter, Beverly, MA, USA), USER Enzyme(NEB, Ipswich, MA, USA), and adaptor-ligated cDNA at 37°C for 15 minutes and subsequently at 95°C for 5 minutes. Then,polymerase chain reaction was performed using the Phusion High-Fidelity DNA polymerase, 10 μM Universal polymerase chain reaction primers, and 10 μM Index (X) Primer. The Agilent Bioanalyzer 2100 system was employed to assess the purification and library quality. The cBot Cluster Generation System using the TruSeq PE Cluster Kit v3-cBot-HS (Illumina,San Diego, CA, USA) was used to yield the cluster, and the Illumina HiSeq2000 platform by Novogene Bioinformatics Technology Co., Ltd. (Beijing, China) was used to evaluate the sequences. Finally, paired-end reads were produced and the clean reads were obtained by removing the adapter (forward:5′-AGA TCG GAA GAG CAC ACG TCT GAA C-3′; reverse: 5′-AGA TCG GAA GAG CGT CGT GTA GGG A-3′) and the Poly-N and low-quality reads (Q < 20) from the raw reads. The read counts, Q10, Q20, Q30, GC base ratio, and average read length of the clean reads were also calculated.
miRNA library construction and sequencing
One microgram of total RNA was used for the construction of the cDNA library using the TruSeq Small RNA Sample Prep Kits (Illumina) according to the manufacturer’s protocol. Next,the Illumina HiSeq 2500 at the LC-BIO (Hangzhou, China) was employed for the single-end sequencing according to the manufacturer’s recommendations. Finally, the clean reads were obtained by Cutadapt V1.14 to remove the 3′-adapter(5′-TGG AAT TCT CGG GTG CCA AGG AAC TC-3′) that controls the length between 17 and 35 bp. Trimmomatic V0.36 was used to delete the low-quality reads (Q < 20), and Blast V2.6.0 was used to remove the RNA families (ribosomal RNA, transfer RNA, small-nuclear RNA, and small-nucleolar RNA) and repeats using comparison conditions that were defined as a gapopen equal to zero, e-value less than 0.01, and mismatch less than 1. To identify novel miRNAs, miRbase V21.0 was used to screen for known miRNA to make a prediction. The unmapped sequences were further searched using miRDeep2 V2.0.0.8,and the mouse reference genome (http://asia.ensembl.org/index.html) was used to distinguish novel miRNAs and predict their secondary structure (Friedl?nder et al., 2012).
Data analysis
Raw data were analyzed using the Empirical Analysis of Digital Gene Expression Data (edgeR) in R. The frequency of the miRNA counts was normalized as reads per million to analyze the expression pattern of miRNAs between APP/PS1 mice and WT control mice. The expression difference was evaluated using Student’st-test. Both a fold change greater than 2.0 and aP-value less than 0.05 were considered as standards to distinguish aberrant miRNAs and mRNAs that were significantly dysregulated between the APP/PS1 mice and WT controls at different age groups. The pheatmap program was also used to visualize the hierarchical clustering of significantly different miRNA expression between APP/PS1 mice and WT mice.
miRNAs target prediction
The known miRNAs were assessed by percent identity between human and mouse species, and the novel miRNAs were evaluated by the miRDeep2 score. In this study, we selected the known miRNAs of a percent identity greater than or equal to 80% and novel miRNAs with a miRDeep2 score greater than or equal to 4 for further analysis. Targetscan, Tarbase, and miRanda were used to search for the potential target genes(Riffo-Campos et al., 2016). The parameters of miRanda were set as single-residue-pair match scores greater than or equal to 150 and ?G less than or equal to -30 kcal/mol.
Gene ontology and Kyoto Encyclopedia of Genes and Genomes pathway analysis
Gene ontology (GO) analysis was performed to explain the molecular mechanism of AD through the molecular function, cellular component, and biological process domains.The Kyoto Encyclopedia of Genes and Genomes (KEGG)pathway enrichment analysis was performed to define the signaling pathway that was associated with the target genes of aberrantly expressed miRNA. The mRNAs-GO-network and mRNAs-KEGG-network were constructed according to the information retrieved from these preliminary analyses.Cytoscape was used to visualize the interaction map between miRNAs and mRNAs. An integrated regulatory diagram was also constructed.Additional Table 2shows the detailed information regarding the software used in this study.
Results
RNA sequencing and analysis for differentially expressed mRNAs in APP/PS1 mice
A total of 24 mRNA relevant cDNA libraries and sequencing were constructed to elucidate the pathogenesis of AD at the transcriptional level, based on a comparative analysis between the WT and APP/PS1 mouse cortex at 1 month, 3 months, 6 months, and 9 months of age. We acquired a mean of 47,675,430 total mRNA clean reads with a percentage of quality value greater than 20 (Q20) and a base ratio higher than 98.59% (Additional Tables 3and4). Subsequently, a more in-depth genome analysis showed that over 96.4% of the clean reads were mapped with the reference genome(Additional Table 5). A total of 51,732 genes expressed in the cerebral cortex of the APP/PS1 transgenic mice were discovered, and, among them, 129 were significantly dysregulated. They were divided into 78 up-regulated and 51 down-regulated genes in the APP/PS1 mouse cortex as compared with the WT control mice at different ages(Additional Table 6), which were potentially involved in AD. The top 10 genes with a significant fold-change in their expression were constructed and represented by upregulated laminin receptor 1 (Lamr1-ps1), S100 calcium-binding protein A8 (S100a8), S100 calcium-binding protein A9 (S100a9),cystatin F (Cst7), chemokine (C-C motif) ligand 3 (Ccl3),AC147560.1, andGm22133, and down-regulatedGm27505,histone cluster 2- H2aa1 (Hist2h2aa1), and mitochondrially encoded transfer RNA serine 2 (mt-Ts2) in APP/PS1 mice compared with the WT control mice (Table 1).
Identification of differentially expressed miRNA in APP/PS1 mice
A total of 24 miRNA relevant cDNA libraries were also constructed, and a mean of 681,080 clean reads after deduplication with a Q20 higher than 97.57% were obtained(Additional Table 7). The clean reads from the 24 miRNAspecific cDNA libraries were aligned using miRBase V21.0 and miRDeep2 V2.0.0.8, which allowed us to identify 1915 known miRNAs and 371 novel miRNAs in the APP/PS1 mouse cortex in contrast to the correspondent control ones (Additional Tables 8and9). Among these known and novel miRNAs, a further evaluation revealed that 68 miRNAs were significantly dysregulated in the APP/PS1 mouse cortex compared with that of the correspondent control mice (Figure 1). Among these significantly dysregulated miRNAs, 6 were increased and 5 were decreased in the APP/PS1 mouse cortex of the 1 month old group. Five miRNAs were significantly increased and eight were significantly decreased in the APP/PS1 mouse cortex of the 3 months old group. Finally, four miRNAs were increased and five miRNAs were decreased in the APP/PS1 mouse cortex of the 6 months old group, and 24 miRNAs were increased and eight miRNAs were decreased in the APP/PS1 mouse cortex of the 9 months old group. Twenty-five significantly expressed miRNAs were novel, and this was the first time they have been identified in the cortex of APP/PS1 mice; these results thus reveal new potential biomarkers that are involved in the pathological process of AD (Table 2). The miRNA-10a-5p was down-regulated in the APP/PS1 mice both at 1 month and 3 months of age, and miRNA-706 was decreased in the APP/PS1 mice at 3 and 6 months of age. Furthermore, novel mature miR-80 and novel mature miR-7 were decreased in the APP/PS1 mice at 6 and 9 months old. Interestingly, novel mature miR-3 was reduced in the 3-month-old APP/PS1 mice, but increased in the 9-month-old APP/PS1 mice (Figure 2). The heat map revealed the expression pattern of the significantly changed miRNAs from 1 to 9 months old, and the hierarchical cluster analysis revealed the clustering of differentially expressed miRNAs in the cerebral samples, as shown inFigure 3.
Prediction of the targets of the significantly expressed miRNAs
The sequence conservation of the differentially expressed miRNA from the cerebral cortex of the APP/PS1 mice was analyzed. Additionally, according to previous reports, a higher miRDeep2 score was taken to indicate a more reliable identification of novel miRNAs (Friedl?nder et al., 2008; Sand et al., 2016). Accordingly, these dysregulated miRNAs were filtered using a percent identity ≥ 80% of known miRNAs and an miRDeep2 score ≥ 4 of novel miRNAs as the cut-off values(Table 2). A total of 8 novel miRNAs and 15 conserved and known miRNAs were obtained, which were further selected to predict potential mRNA targets. A total of 21,322 putative target genes were intersected with these selected miRNAs using the above algorithms. Notably, 12 highly conservedknown miRNAs and 3 novel miRNAs with the corresponding target genes were tightly connected with AD pathogenesis,including miR-10a-5p, miR-10b-5p, miR-96-5p, miR-144-3p, miR-192-3p, miR-211-5p, miR-214-3p, miR-215-5p, miR-223-3p, miR-361-5p, miR-296-5p, miR-451a, novel mature miR-29, novel mature miR-80, and novel mature miR-102.Their target genes were found to be specific and closely related to AD, and includedAPP, beta-secretase 1 (BACE1),ADAM metallopeptidase domain 10 (ADAM10), insulin-like growth factor 1 (IGF1), autophagy related 12 (ATG12), brainderived neurotrophic factor (BDNF), B-cell lymphoma 2(BCL2) apoptosis regulator, and the limb-bud and heart (LBH)regulator of the wingless-type (WNT) signaling pathway (Table 3). Subsequently, the miRNA-mRNA interaction was merged to construct a network. mRNAs and miRNAs were defined as the network nodes, and an edge was added between nodes that interacted with each other. This intersected network contained 25 mRNAs and 15 miRNAs. Thus, based on the interaction network (Figure 4), we constructed the cerebral miRNA-mRNA network associated with the pathological AD process.
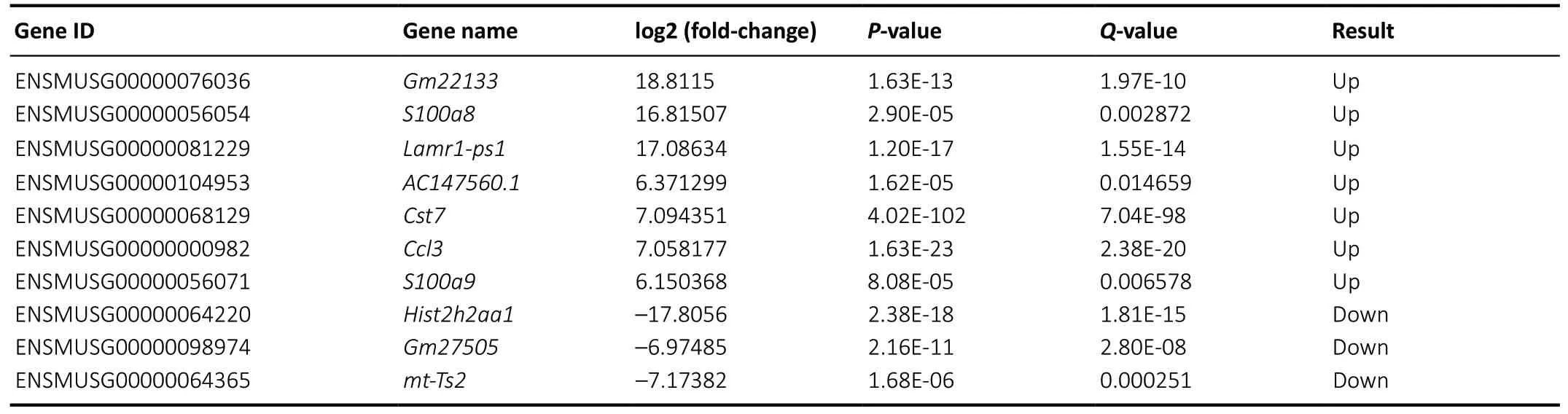
Table 1 |Top 10 of the most significantly dysregulated mRNAs in the AD mouse cortices at 1, 3, 6, and 9 months of age
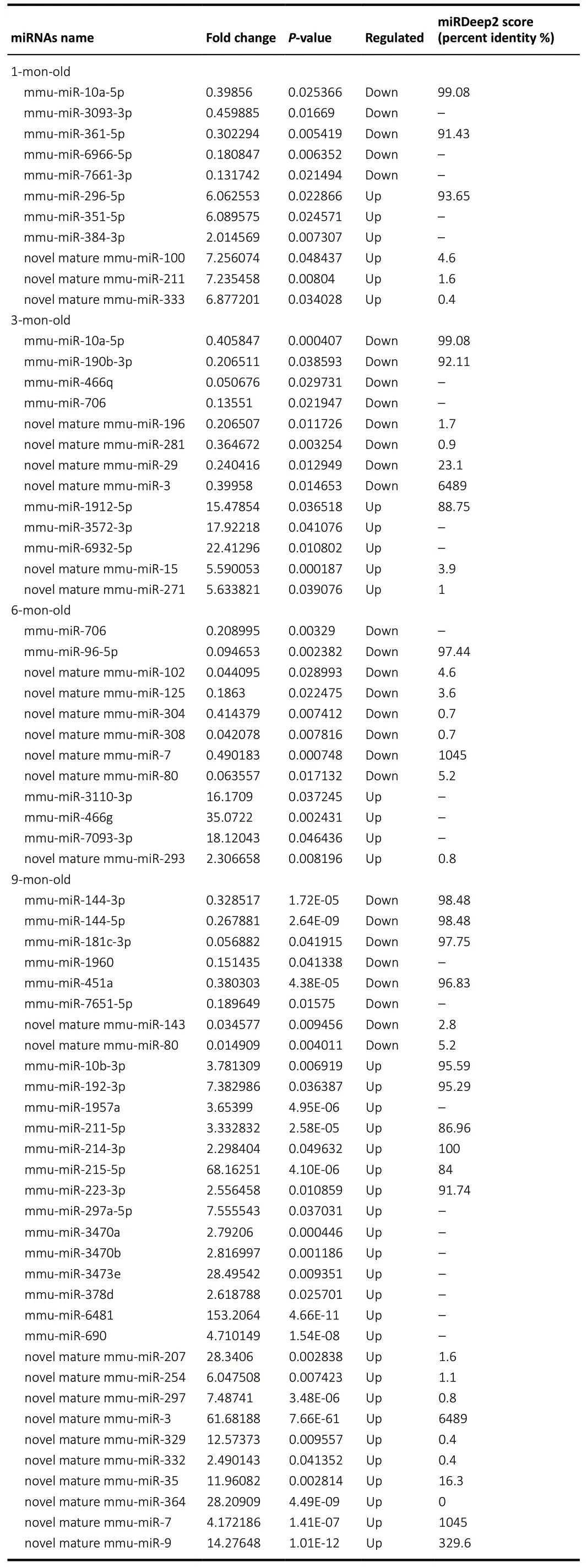
Table 2 |Dysregulated miRNAs in the AD mouse cortices (vs. WT control mice) at 1, 3, 6, and 9 months of age
GO and KEGG pathway analysis of the dysregulated miRNAs
The dysregulated miRNAs led to a change of gene expression,which resulted in activation of the pathological signaling pathway that causes AD.Figures 5and6display the top 10 GO enrichment domains of the target genes of the significantly dysregulated miRNAs; the biological process terms of GO enrichment were involved in gene silencing by miRNAs development, transcription DNA-template development,nervous system development, and phosphorylation development. Moreover, identification of the top ten cellular components indicated that the differentially expressed miRNAs were located at the synapse, in the nucleus, and on the cell membrane. Furthermore, the molecular function terms revealed a role of the top ten molecular functions in protein phosphatase binding, actin binding, and activating transcription factor binding. The KEGG pathway analysis also revealed an involvement of mitogen-activated kinase protein (MAPK), phosphatidylinositol 3-kinase-protein kinase B (PI3K-AKT), mechanistic target of rapamycin kinase (mTOR),forkhead box O (FOXO), axon guidance, olfactory transduction,pancreatic secretion, neuroactive ligand-receptor interaction,and autophagy in AD, as shown inFigures 7and8. The GO top enrichment network (Additional Figure 3) and pathway top enrichment network (Additional Figure 4) were established to clarify the potential regulatory mechanism in the pathogenesis of AD, as well as the interaction between miRNAs and mRNAs.

Table 3 | Significantly changed miRNAs and their predicted target genes,including miRNAs name, sequence, and target genes
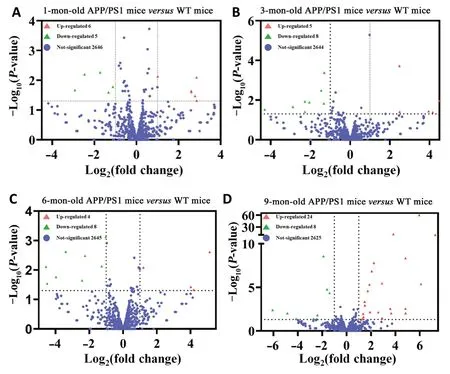
Figure 1|Volcano plots of the differentially expressed miRNAs in the APP/PS1 mouse cortices at 1 (A), 3 (B), 6 (C), and 9 months (D) of age, in contrast to the corresponding control ones.
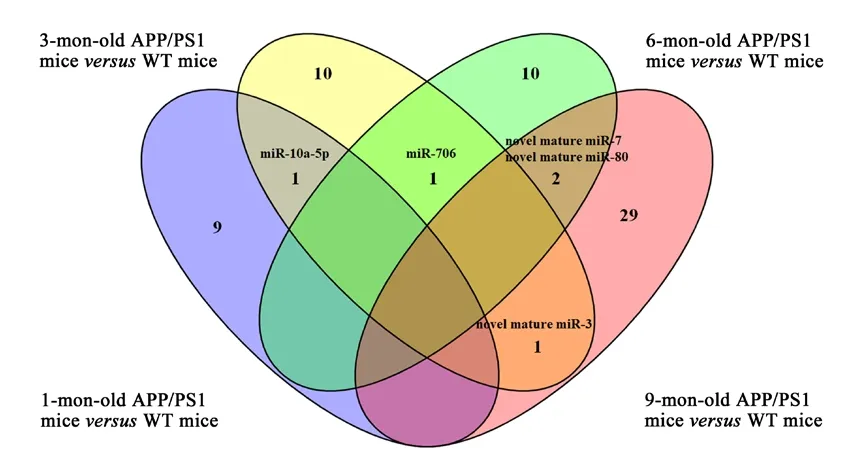
Figure 2|Venn diagram of the aberrantly changed miRNAs in the APP/PS1 mice at 1, 3, 6, and 9 months of age.
Discussion
No cure for AD is available, and there is not yet an effective treatment to inhibit or slow its progression; however, some options are available to treat the symptoms of AD. Growing evidence has indicated that the best way to avoid AD or delay symptom onset effectively is prevention and early diagnosis, rather than treatment (Dolgin, 2018). To this aim,increasingly efficient and accurate sequencing technology has been used to help identify biomarkers and differentially expressed genes that are related to the pathogenesis of AD.miRNAs are negative regulators of genes, widely distribution throughout body tissues, and easy to detect, and regulate genes at a transcriptional and post-transcriptional level to reduce gene silencing. Increasing evidence has revealed that the dysregulation and alteration of miRNAs can lead to the occurrence of AD (Silvestro et al., 2019). The identification of molecular biomarkers represented by aberrantly expressed miRNAs and mRNAs could facilitate the diagnosis and prognosis of AD before the onset of the symptoms; thus, they could be potential drug targets.

Figure 3|Hierarchical cluster analysis of differentially expressed miRNAs in APP/PS1 mice at 1 (A), 3 (B), 6 (C), and 9 months (D) of age.
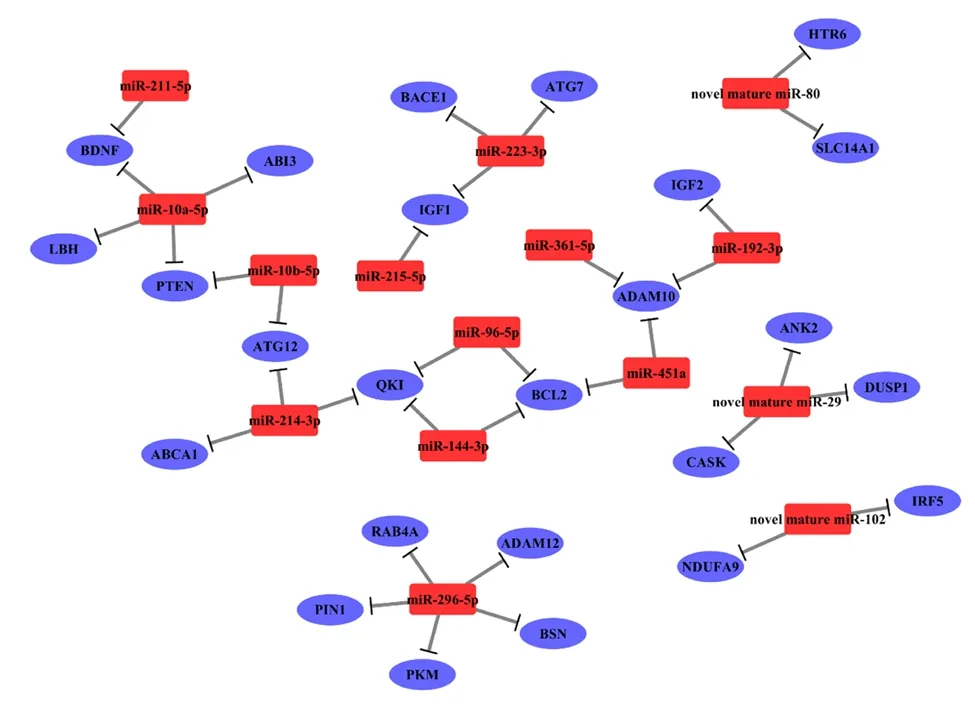
Figure 4|The miRNA-mRNA network of the significantly expressed miRNAs and their corresponding targets genes in the APP/PS1 mouse cortices at 1, 3, 6, and 9 months of age in contrast to the corresponding control ones.
APP/PS1 transgenic mice simulate human AD-like senile plaques symptoms by overproduction of Aβ, the expression of which increases with age (Sun et al., 2019). Thus, these mice are widely used to investigate the occurrence and development of AD. The cerebral cortex and hippocampus regulate learning and memory (Rakic et al., 1994). The entorhinal, prefrontal, and temporal areas are cerebral regions that are affected during the early stages of AD (Gaffan, 2002),and play an indispensable role in the cortex-hippocampal circuit for memory and learning formation. Therefore, the integrated cerebral cortex was used to identify aberrant genes and biological pathways related to neuropathological changes in AD.

Figure 5|Top 10 GO enrichment analysis of the target genes of the down-regulated miRNAs in the APP/PS1 mouse cortices at 1 (A), 3 (B), 6 (C), and 9 months (D) of age.
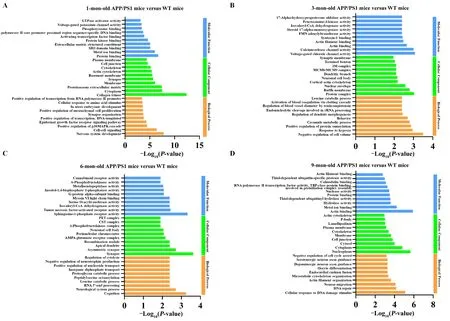
Figure 6|Top 10 GO enrichment analysis of the target genes of the significantly increased miRNAs in the APP/PS1 mouse cortices at 1 (A), 3 (B), 6 (C),and 9 months (D) of age.
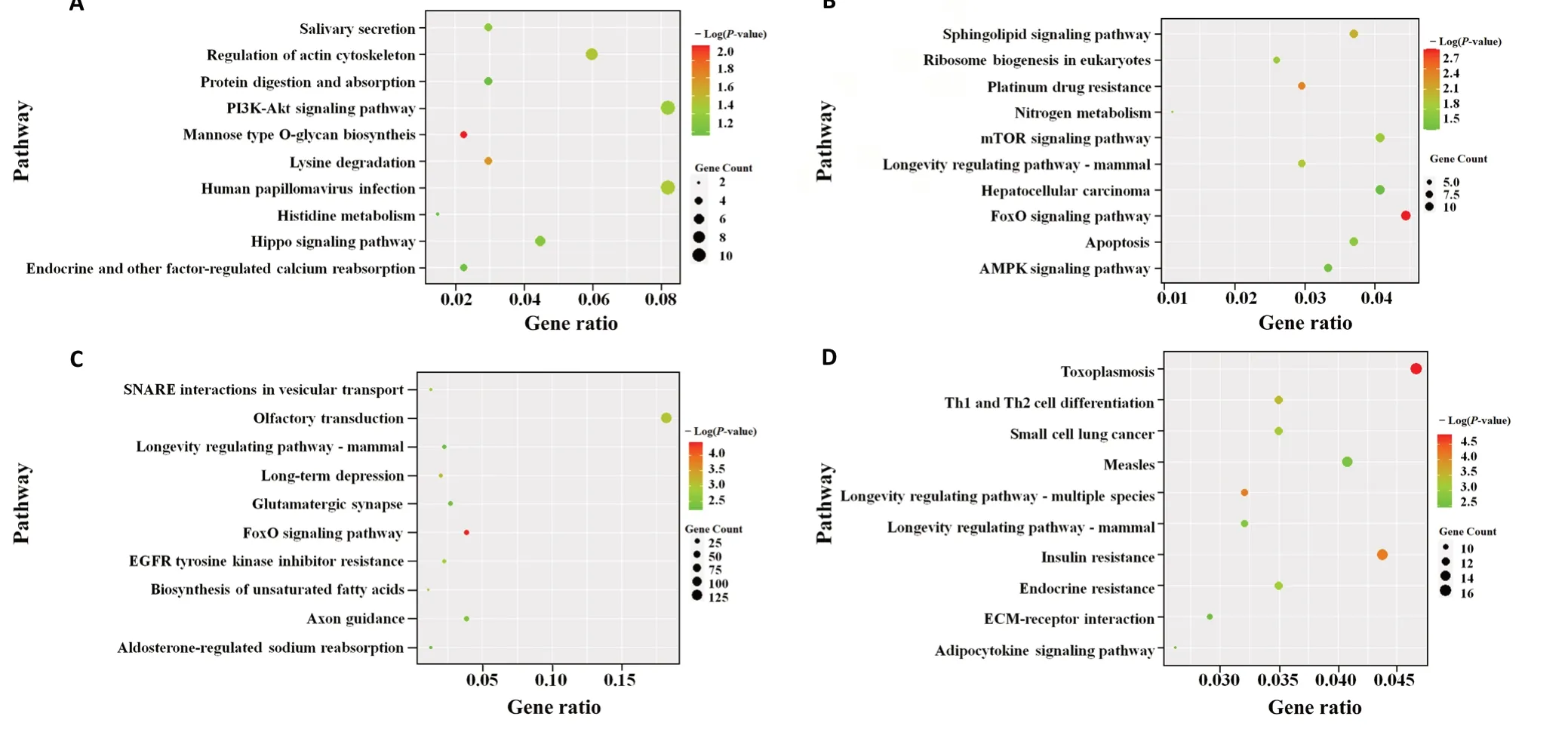
Figure 7|Top 10 KEGG pathway analysis of the target genes of the significantly decreased miRNAs in the APP/PS1 mouse cortices at 1 (A), 3 (B), 6 (C),and 9 months (D) of age.
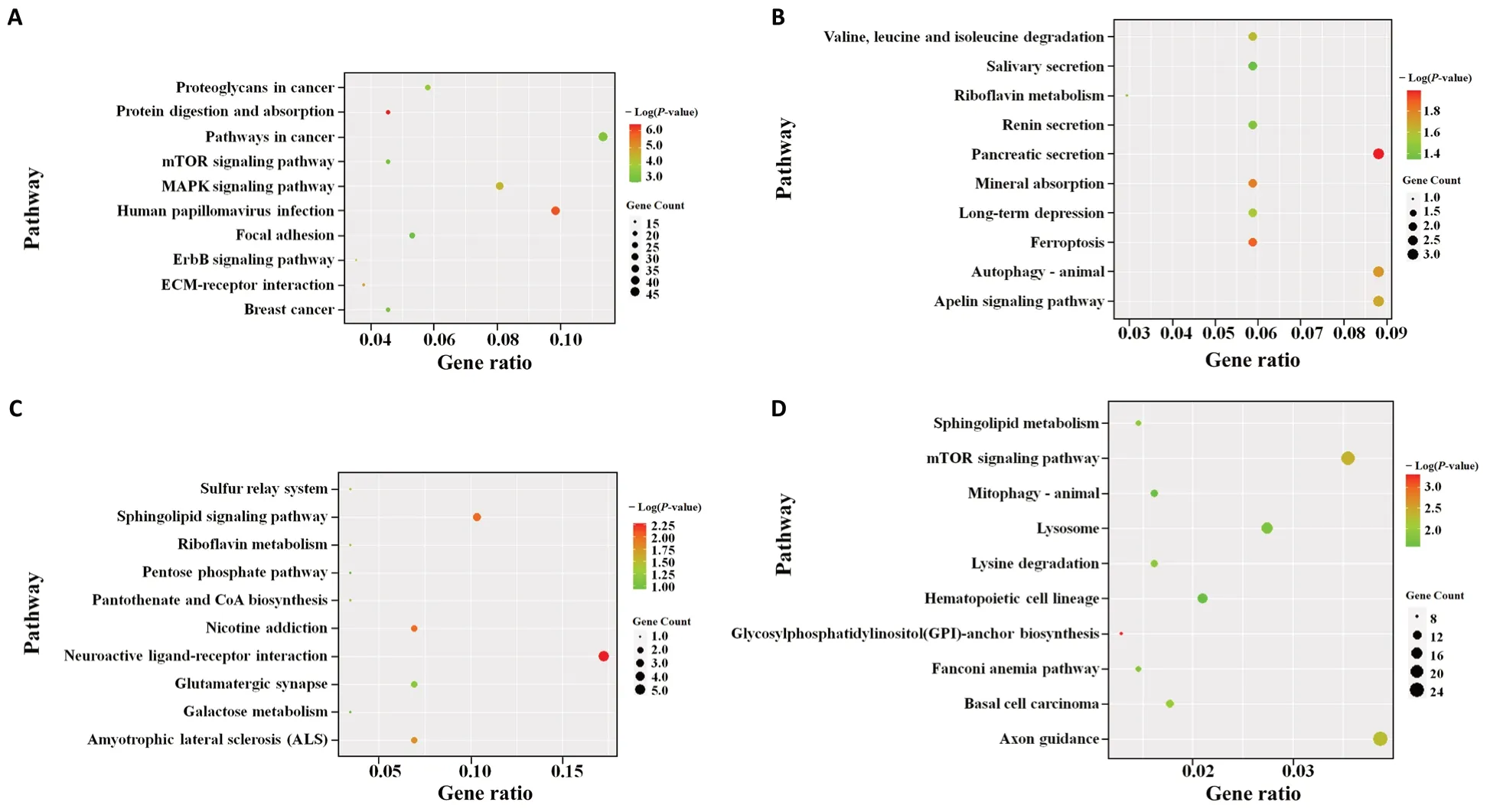
Figure 8|Top 10 KEGG pathway analysis of the target genes of the significantly increased miRNAs in the APP/PS1 mouse cortices at 1 (A), 3 (B), 6 (C),and 9 months (D) of age.
In this study, 129 significantly changed genes were identified in APP/PS1 transgenic mice, consisting of 78 up-regulated and 51 down-regulated genes. The top ten most significantly changed genes were up-regulated, such asLamr1-ps1,S100a8,S100a9,Cst7,Ccl3,AC147560.1, andGm22133,while the down-regulated ones wereGm27505,Hist2h2aa1,andmt-Ts2. However, to the best of our knowledge, this is the first report to linkLamr1-ps1,AC147560.1,Gm22133,Gm27505,Hist2h2aa1, andmt-Ts2genes with AD. Among the genes known to be altered in AD,S100a8has been found to be up-regulated with age (Lodeiro et al., 2017), which was corroborated by our high-throughput results. S100a8 also plays a crucial regulatory role in inflammatory processes that occur prior to Aβ over-aggregation resulting from the positive feedback of Aβ production, which leads to memory and learning impairment (Lodeiro et al., 2017). Similarly,S100a9 drives an amyloid-neuroinflammatory cascade in the precursor phase of AD (Wang et al., 2018). Microgliaassociated amyloidosis may play a pivotal role in AD pathology(Ofengeim et al., 2017). In AD and similar pathologies, Cst7 encodes cystatin F, which acts as a cellular marker of diseaseassociated microglia, and is responsible for the uptake of extracellular Aβ through autophagic and lysosomal pathways.Thus, Cst7 indirectly inhibits excessive Aβ deposition.
A total of 68 miRNAs were significantly dysregulated, including 39 up-regulated and 29 down-regulated miRNAs. For the first time, this study identified 25 novel aberrant miRNAs in the cerebral cortex of APP/PS1 mice, which were dysregulated at different stages of AD. Among these miRNAs, miR-10a-5p was down-regulated in the APP/PS1 mouse cortex at 1 month and 3 months old, which indicates that it is involved in the early pathogenesis of AD. Moreover, miR-10a-5p has an anti-inflammatory effect by simultaneously binding to two target genes, the mitogen-activated protein kinase kinase 7 and the β-transducin repeat-containing gene, to inhibit proinflammatory cytokines and chemokines in endothelial cells(Fang et al., 2010). Thus, this study hypothesized that miR-10a-5p exerts a neuroprotective effect in the pathogenesis of AD by alleviating the inflammatory response. Unlike the action of miR-706 as a pro-cell death miRNA that is detected in endoplasmic reticulum-mediated diseases (Wang et al.,2020), miR-706 in our study was down-regulated in APP/PS1 mice at 3 and 6 months of age; this suggests, for the first time,that miR-706 plays a regulatory role in the intermediate stage of AD. Furthermore, RNA sequencing identified novel mature miR-80, novel mature miR-7, and novel mature miR-3, which were reduced in the advanced stage of AD.
The miRNA-mRNA network has an important influence on complex gene expression. This study constructed an miRNAmRNA network based on 12 highly conserved miRNAs and three reliable novel miRNAs that were significantly expressed.Interestingly, the conserved miRNAs, such as miR-10a, miR-10b, miR-361-5p, miR-296-5p, miR-144, miR-451, miR-192,miR-214, miR-215, miR-223, miR-190b, and miR-181c, have also been reported to be differentially expressed in the human anterior cingulate gyrus and motor cortex (Nelson et al.,2018). The corresponding target genes were strongly related to the etiopathogenesis of AD, and includedAPP,BACE1,ADAM10,ADAM12, KH domain-containing RNA binding (QKI),BDNF, phosphatase and tensin homolog (PTEN),BCL2,IGF1,andLBH, which are key regulatory genes and/or enzymes in Aβ and Tau biosynthesis and transportation, synaptic dysfunction,neuronal apoptosis, autophagy, and inflammation (Malinin et al., 2005; Farnsworth et al., 2016; Yamaguchi-Kabata et al.,2018; Silvestro et al., 2019). Assuming that this miRNA-mRNA network covers multiple aspects of AD pathology, these gene regulators might be novel targets in developing additional treatments for AD.
Among the highly conserved miRNAs identified, the differentially expressed miR-10b-5p, miR-214-3p, miR-215-5p,and miR-296-3p play roles in differentiation, cell proliferation,migration, and invasion, which suggests that they could be diagnostic biomarkers in a variety of cancer-related diseases(Tao et al., 2019; Wang et al., 2019b; Cho et al., 2020; Liu et al., 2020; Wu et al., 2020). However, to the best of our knowledge, the discovery of their action in AD pathology in the present study is novel. Moreover, miR-10b-5p has an anti-apoptotic effect by directly binding to the 3′-UTR of PTEN, as demonstrated by the regulatory function in neuron apoptosis and ER stress via activation of the PI3K/AKT pathway in AD (Cui et al., 2017; Wu et al., 2019). Hence, miR-10b-5p could be involved in neuroprotection and ER regulation through pairing with the 3’-UTR of PTEN. miR-223-3p is another conservative miRNA that participates in various neurodegenerative diseases, and has considerable potential as a non-invasive biomarker in identifying AD, Parkinson’s disease, and mild cognitive impairment (Mancuso et al., 2019).
Some novel miRNAs, such as novel mature miR-29, novel mature miR-80, and novel mature miR-102, were identified for the first time in our AD model. The target genes of novel mature miR-29 are dual specificity phosphatase 1 (DUSP1),calcium/calmodulin dependent serine protein kinase(CASK), and ankyrin 2 (ANK2), which play crucial roles in AD associated-pathogenesis (Leandro et al., 2018; Higham et al., 2019; Silva et al., 2020). Given that the ANK2 gene contributes to longevity, cognition, and neurotransmitter conduction (Jenkins et al., 2015; Michalak et al., 2017), it is possible that the novel mature miR-29-ANK2 interaction is associated with multiple pathological mechanisms in AD.The predicted genes of the down-regulated novel mature miR-80 were the 5-hydroxytryptamine receptor 6 (HTR6)and solute carrier family 14 member 1 (SLC14A1). The 5-HT6 receptor polymorphism (C267T) of HTR6 participates in the susceptibility to late-onset AD, and SLC14A1 is modified in neurodegenerative diseases such as AD (Kan et al., 2004;Recabarren and Alarcón, 2017). Novel mature miR-102 exerts neuroprotective effects by targeting NADH: ubiquinone oxidoreductase subunit A9 (NDUFA9) and interferon regulatory factor 5 gene (IRF5) (Zhu et al., 2016; Adav et al.,2019), which are involved in M2 microglia activation through the inhibition of neuroinflammation and consequently better cognition. Thus, these novel miRNAs might be promising biomarkers of AD.
Concerning the GO enrichment analysis results, the dysregulated miRNAs from the APP/PS1 mice played a crucial role in the transcription and post-transcription stages. The target genes of the aberrant miRNAs are primarily involved in the biological processes of neuron differentiation,transportation process, and apoptosis. Our pathway enrichment analysis revealed the involvement of MAPK, FOXO,mTOR, and PI3K-AKT pathways, neuroactive ligand-receptor interaction, and autophagy, as well as pathological processes already implicated in the pathogenesis of AD, such as tau phosphorylation, biosynthesis of APP, loss of neurons, and the neuroinflammatory reaction. These miRNAs and potential target molecules should be further investigated in future experiments.
Current AD therapies are still ineffective in preventing and curing this disease, and more effective interventions are therefore urgently required. Part of the solution, which could aid the development of both an effective diagnostic method and AD therapy, is the analysis of gene expression modification linked to AD for a better-individualized control of AD patients (Ansari et al., 2017). However, one limitation of this work is the high false positive rate derived from algorithms and analysis software. This limitation means that different animal models, as well as serum from patients with AD, will be needed to assess the validity of our sequencing results using quantitative polymerase chain reaction, western blots, and immunohistochemistry.
In conclusion, this study revealed the aberrant miRNA and mRNA expression profile that contributes to the pathogenesis of AD, and identified significantly expressed mRNAs, miRNAs,and miRNA potential target genes. This work therefore puts forward representative novel therapeutic targets and promising biomarkers in the diagnosis of AD.
Author contributions:Concepts: RL; study design: ZRL, RL; definition of intellectual content: ZRL; literature search: LZ, HLJ, GMA; experiment implementation, data analysis and manuscript preparation: LZ, HLJ;manuscript editing: GMA, RL; manuscript review: GMA, ZRL, RL. All authors read and approved the final manuscript.
Conflicts of interest:The authors declare no conflict of interest in this research.
Financial support:This study was supported by the National Natural Science Foundation of China (General Program), No. 81673411; the United Fund Project of National Natural Science Foundation of China, No.U1803281; Young Medical Talents Award Project of Chinese Academy of Medical Sciences, No. 2018RC350013; and Chinese Academy of Medical Sciences Innovation Project for Medical Science, No. 2017-I2M-1-016 (all to RL). The funding sources had no role in study conception and design,data analysis or interpretation, paper writing or deciding to submit this paper for publication.
Institutional review board statement:This study was approved by Ethical Committee of the Institute of Medicinal Biotechnology, Beijing,China (approval No. IMB-201909-D6) on September 6, 2019.
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement:Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix,tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Additional files:
Additional Figure 1: APP/PS1 mice showing learning and memory deficits in the Morris water maze test.
Additional Figure 2: Purity evaluation of total RNA in 1% agarose gel electrophoresis.
Additional Figure 3: The 1- (A), 3- (B), 6- (C), and 9-month-old (D) Gene Ontology enrichment top network.
Additional Figure 4: The 1- (A), 3- (B), 6- (C), and 9-month-old (D)pathway enrichment top network.
Additional Table 1: Concentration of total RNA by Spark 20M multimode microplate reader.
Additional Table 2: Website information regarding the software used in this study.
Additional Table 3: Quality assessment of mRNA sequencing reads including total read counts, total bases counts, average read length, N bases count, N bases ratio, GC bases count, and GC bases ratio.
Additional Table 4: Quality assessment of mRNA sequencing reads including Q10/Q20/Q30 bases count and Q10/Q20/Q30 bases ratio.
Additional Table 5: Summary of the genome mapping analysis in the mRNA sequencing, including total reads, total mapped, multiple mapped,and uniquely mapped.
Additional Table 6: Differentially expressed genes in the APP/PS1 mouse cortices at 1, 3, 6, and 9 months in contrast to the same age of control mice.
Additional Table 7: Summary of the miRNA sequencing reads.
Additional Table 8: Summary of the known miRNA prediction using miRBase and miRDeep2 in the APP/PS1 mouse cortices at the four tested ages.
Additional Table 9: Summary of the novel miRNA prediction using miRBase and miRDeep2 in the APP/PS1 mouse cortices at the four tested ages.
- 中國神經(jīng)再生研究(英文版)的其它文章
- Pharmacological interventions targeting nuclear factor-kappa B signaling in multiple sclerosis
- Corrigendum
- Proteolysis targeting chimera technology: a novel strategy for treating diseases of the central nervous system
- Notice of Retraction
- Luteolin delays photoreceptor degeneration in a mouse model of retinitis pigmentosa
- Fluoxetine, a selective serotonin reuptake inhibitor used clinically, improves bladder function in a mouse model of moderate spinal cord injury