
Base editing: a brief review and a practical example
2021-04-09 12:47:40DongwookChoeKiranMusunuru
Dongwook C. Choe, Kiran Musunuru
Division of Cardiology and Cardiovascular Institute, Department of Medicine, Department of Genetics, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA 19104, USA.
Abstract Genome editing has undergone rapid development in recent years, yielding new approaches to make precise changes in genes. In this review, we discuss the development of various adenine and cytosine base-editing technologies, which share the ability to make specific base changes at specific sites in the genome. We also describe multiple applications of base editing in vitro and in vivo. Finally, as a practical example, we demonstrate the use of a cytosine base editor and an adenine base editor in human cells to introduce and then correct a prevalent mutation responsible for hereditary tyrosinemia type 1.
Keywords: base editing, CRISPR, genetics, genome editing, mutation
Introduction
Precision genome editing is a growing field with industrial, agricultural, and biomedical applications.One of the dominant genome-editing systems available today is clustered regularly interspaced short palindromic repeats (CRISPR)-CRISPR-associated protein 9 (Cas9). Through the use of a guide RNA(gRNA) with a sequence homologous to that of a sequence of DNA in the target genome (known as the protospacer) adjacent to a specific protospaceradjacent motif (PAM) in the DNA, Cas9 can be used to create a double-strand break (DSB) at the targeted sequence[1]. Non-homologous end joining at DSBs can be used to create indels and knock out genes at genetic loci; likewise, homology-directed repair (HDR) can be used, with an introduced template DNA, to insert genes or modify the targeted sequence[2]. A variety of Cas9-based tools have been developed in recent years,including tools that methylate DNA, recognize the broader sequence space, or create single-strand nicks.In this review, we describe Cas9-based tools that can generate specific point mutations in DNA without needing to create DSBs: base editors. We describe published examples of their usein vitroandin vivo,and we then present a specific application of such tools.
Cytosine base editing
In 2016, Komoret aldescribed the use of CRISPRCas9 to convert a cytosine base to a thymine base without introducing a template DNA strand[3]. After the cytidine deaminase domain of rat APOBEC1 was fused to the N-terminus of catalytically-dead Cas9(dCas9) using the linker XTEN (resulting in a fusion protein called base editor 1, or BE1), conversion of cytosine to uracil was observed between position 4 and position 8 within the 20-nt protospacer region of DNA (or, to express it a different way, 13thto 17thnucleotides upstream of the PAM). Of note, any cytosine base within this "window" was amenable to editing, resulting in varied outcomes depending on which cytosines were edited and how many they were.After DNA replication or repair, each uracil was replaced by thymine, completing the C→T base editing[3]. The next version of base editor (BE2)incorporated a uracil glycosylase inhibitor fused to the C-terminus of dCas9 to help inhibit base excision repair of the uracil bases resulting from the cytidine deaminase activity (which otherwise would act to restore the original cytosine bases); this improved the efficiency of C→T base editing. The final version,BE3, used a Cas9 nickase rather than dCas9; the nickase cut the unedited strand opposite the edited C→T bases, stimulating the removal of the opposing guanidine through eukaryotic mismatch repair (Fig. 1).BE2 and BE3 base editing was observed in both human and murine cell lines. The specificity of base editing has been further improved through the addition of mutations to the Cas9 nickase[4]; in a similar fashion, Cas9 has been mutated to narrow the width of the editing window from approximately 5 nucleotides to as little as 1-2 nucleotides[5].
An alternative cytosine base editing platform was described by Nishidaet alin the same year[6]. By linking the activation-induced cytosine deaminase(AID) domain PmCDA1 to dCas9 (Target-AID), they were able to demonstrate targeted C→T base editing in yeast. Furthermore, an alternative C→T editing strategy was also demonstrated without fusing a deaminase domain to Cas9; instead, a SH3 (Src homology 3) domain was added to the C-terminus of dCas9 while a SHL (SH3 interaction ligand) was added to PmCDA1[6]. Optimization of efficiency was achieved through the use of a Cas9 nickase rather than dCas9, similar to the strategy employed by Komoret al[3]. Further, a uracil DNA glycosylase inhibitor was added to enhance base editing in the mammalian CHO cell line. The resulting platform was able to consistently edit bases within 3 to 5 bases of the 18thnucleotide upstream of the PAM sequence.
A distinct cytosine base editing platform was reported by Liet alin 2018[7]. Unlike the former two editors, Liet alused Cpf1 (also known as Cas12a)instead of Cas9 as the RNA-guided endonuclease.Catalytically-inactive Cpf1 was fused to APOBEC1(dLbCpf1-BE0), leading to C→T conversion in a human cell line. While the Cas9 base editor variants BE3 and Target-AID recognize the PAM sequence NGG, dLbCpf1-BE0 recognizes the T-rich PAM sequence TTTV. Although base editing was observed between positions 8 and 13 of the protospacer sequence with dLbCpf1-BE0, the introduction of additional mutations into Cpf1 was able to reduce the window to positions 10 to 12[7]. However, narrowing of the base editing window correlated with a decrease in editing efficiency.
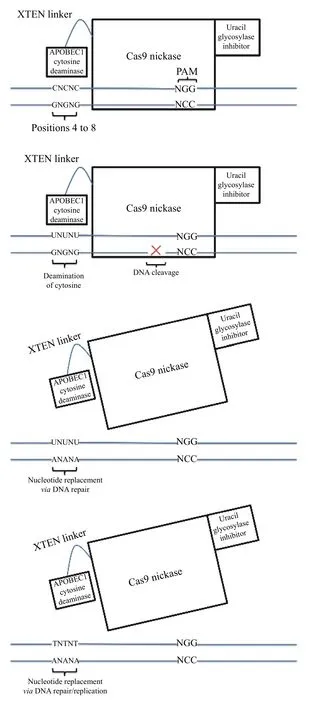
Fig. 1 Schematic of mechanism of action of BE3, a cytosine base editor. A: adenine; C: cytosine; G: guanine; T: thymine;U: uracil; N: any base
Cytosine base editing is not wholly predictable;indels can occur at the target site, albeit at lower frequencies than those observed for C→T editing editors[3,6]. Furthermore, cytosine base editors can occasionally cause C→A or C→G edits rather than the expected C→T edits. To address this problem,Komoret almade three changes to the standard BE3 base editor[8]. The linker between Cas9 nickase and the rat APOBEC1 cytosine deaminase domain was increased from 16 amino acids to 32 amino acids, the linker between the Cas9 nickase and the uracil glycosylase inhibitor was increased from 4 amino acids to 9 amino acids, and a second uracil glycosylase inhibitor was appended to the C-terminus of the new cytosine base editor using another 9 amino acid linkers. This improved cytosine base editor was termed "BE4", in keeping with the BE(x) naming convention. Upon comparison to BE3 in HEK 293T cells across multiple loci, BE4 showed a 1.5-fold increase in C→T editing over BE3, whereas C→A and C→G edits decreased by 2.3 folds; indels were also reduced by 2.3 folds. A variant using Cas9 nickase derived fromStaphylococcus aureus(rather thanStreptococcus pyogenes, which is the most commonly used version) was also created, showing an increase in editing efficiency by 1.4 folds and a decrease in non-C→T edits by 1.8 folds over the standard BE3[8]. This variant was named SaBE4.Furthermore, in light of the hypothesis that Gam protein could protect against indels by binding to the ends of DSBs, Gam was fused to the N-termini of BE4, SaBE4, BE3, and SaBE3. Frequencies of indels and non-C→T edits for the Gam fusion proteins were universally lower than those for their non-Gam counterparts[8].
Adenine base editing
Cytosine base editors permit the introduction of C→T changes in the coding sequences of genes; if directed to the opposite (non-coding) strand, they can also introduce G→A coding changes. These represent just 2 out of 12 possible types of base edits. In principle, the existence of adenine deaminase enzymes that convert adenine to inosine, which is then replaced with guanine, would permit A→G edits (resulting in either A→G or T→C coding changes, depending on which strand was targeted). Unfortunately, no natural adenine deaminases that act on DNA are known to exist. However, by fusingEscherichia coliadenine tTNA deaminase TadA (ecTadA) to dCas9, Gaudelliet alhoped to demonstrate adenine base editing capability on targeted DNA sequences[9]. Mutagenesis of the ecTadA domain in conjunction with selection for editing activity revealed that A106V and D108N mutations yielded a base editor capable of editing adenine to guanine in DNA. Successive rounds of mutagenesis, selection, and additional improvements yielded ABE7.10 (Fig. 2), an A→G base editor capable of reaching up to 50% efficiency in human cells with low indel rates[9]. The editing window ranges from positions 4 to 7 in the protospacer sequence for ABE7.10 to positions 4 to 9 with earlier versions of adenine base editors (ABE7.9, ABE7.8,and ABE6.3). Off-target activity by ABE7.10,ABE7.9, and ABE 7.8 in HEK 293T cells appeared to be lower than that caused by standard Cas9 editing,and no ABE-induced editing was detected outside ontarget or off-target protospacers.
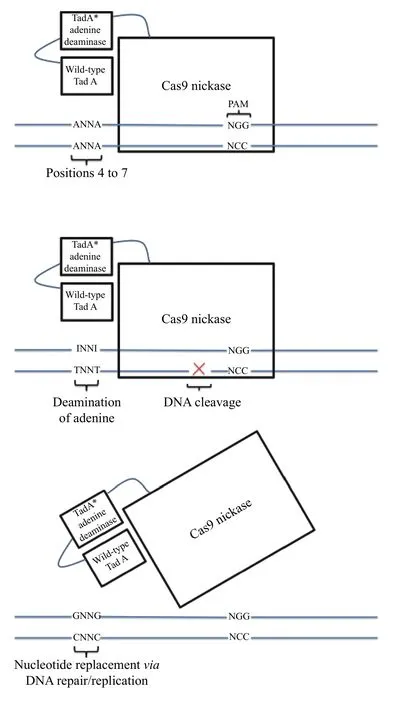
Fig. 2 Schematic of mechanism of action of ABE7.10, an adenine base editor. A: adenine; C: cytosine; G: guanine; I: inosine/hypoxanthine; T: thymine; N: any base
Koblanet alimproved the efficiency of ABE7.10 through modification of nuclear localization signals and codon optimization, yielding a version called ABEmax; a similar approach improved the efficiency of the cytosine base editor BE4[10]. Huanget alperformed further development of both adenine and cytosine base editors to use alternative PAMs and to expand their editing windows, thereby increasing their targeting range[11].
Off-target effects of base editors
With standard CRISPR-Cas9 genome editing, offtarget mutagenesis—i.e., edits at genomic sites other than the intended target site—can occur at sites that share a high degree of sequence similarity with the protospacer sequence encoded in the gRNA. The same has proven to be true of base editors[12-14], although comparisons of Cas9, cytosine base editors, and adenine base editors using the same gRNAs have shown distinct off-target profiles.
Recently, a variety of studies have raised concern about gRNA-independent off-target base editing,caused by the deaminase domain acting in isolation(without the need for engagement of DNA by the Cas9-gRNA complex). Zuoet alcompared off-target single nucleotide variants (SNVs) caused by Cas9,BE3, and ABE 7.10[15]. They injected one cell of a two-cell mouse embryo with one of the genomeediting tools, and after growth of the embryo, they directly compared SNV frequencies between edited and nonedited embryonic cells on embryonic day 14.5. The excess of SNVs in the edited cells was attributed to the genome-editing tool. Although embryos injected with Cas9 or ABE7.10 showed induced SNV rates similar to that of control embryos,embryos injected with BE3 had an SNV rate approximately twenty times higher than that of the control group (243 SNVs per embryo). Approximately 90% of all BE3-associated SNVs were C→T mutations; this bias was not observed in the Cas9 or ABE7.10 experimental groups. SNV formation in the BE3 group was gRNA-independent,i.e., was not linked to protospacer sequence similarity or the presence of PAMs. As a result, the authors hypothesized that the off-target effects were caused by the overexpression of the APOBEC1 subunit. Similar results were found by Jinet alfor cytosine base editing in rice[16].
Studies by Grünewaldet al[17-18]and Zhouet al[19]showed that the gRNA-independent off-target effects are not limited to DNA. RNA sequencing of cells treated with either cytosine base editors or adenine base editors revealed transcriptome-wide off-target editing of RNA. Although the same groups showed that protein engineering of base editors, specifically in the deaminase domain, can reduce gRNA-independent off-target effects on RNA and DNA, the consequences of these off-target effects require further scrutiny,particularly if base editors are to be used for therapeutic purposes.
Potential use of base editors for mutagenesis screens
Although the base editors were intended to be used in a sequence-specific manner, other base-editing strategies exist to create a library of point mutations in a defined location on a genome. Targeted AIDmediated mutagenesis (TAM), developed by Maet al,uses a dCas9 fused to an AID in conjunction with gRNAs to mutate cytosine to the other three bases in loci targeted by the gRNAs[20]. After removing the nuclear export sequence from AID to generate AIDx,over 20% of C bases (or G bases, if targeting the opposite, non-coding strand) in the targeted GFP locus could be modified to another base using TAM. Some bias for C→T or G→A mutations was observed over the other possible base mutations, and a small proportion of A and T bases underwent mutagenesis at the targeted locus. When TAM was applied in the presence of a uracil glycosylase inhibitor, mutation frequencies increased; however, editing became strictly limited to C→T mutations between 12thand 16thnucleotides upstream of the PAM sequence.
Hesset aldetailed an alternative approach towards library creation through the editing strategy called CRISPR-X[21]. By using dCas9 in conjunction with gRNAs bearing MS2 hairpin binding sites, they were able to recruit AID variants fused to MS2 binding protein to mutagenize longer stretches of DNA, rather than being restricted to small nucleotide windows in the protospacer. In particular, one hyperactive variant of AID, AID*Δ, was able to edit bases in GFP between 50 nucleotides upstream and 50 nucleotides downstream of the PAM sequence. Similar to TAM,CRISPR-X showed a preference for C→T and G→A mutations and demonstrated a more limited capacity for A and T base editing.
Potential use of base editors for therapeutic applications
The ability to edit bases directly allows for the disruption of genesin vivowithout the need to create DSBs. For example, Chadwicket alused cytosine base editing to introduce nonsense mutations into thePcsk9gene in adult mice[22]. Delivery of BE3 and a gRNAviaadenoviral vector into the liver caused a reduction in plasma PCSK9 levels by 56%.Furthermore, cholesterol levels in the base-edited mice were reduced by roughly 30%. In subsequent work, cytosine base editing to disrupt theAngptl3gene in adult mice resulted in substantially reduced blood cholesterol and triglyceride levels[23], and cytosine base editing to disrupt thePcsk9gene or theHpdgene in fetal mice resulted in postnatal reduction of cholesterol levels or cure of the disease hereditary tyrosinemia type 1[24].
Correction of disease-causing mutationsviaprecision editing with standard Cas9 genome editing has largely required HDR. Since HDR is limited to cells in the S or G2 phase of mitosis, precision editing of non-mitotic cells is difficult. However, base editors are not reliant on HDR; as demonstrated by Yehet al,the editing of postmitotic cochlear cells in mice is feasible with cytosine base editor BE3[25]. By injecting BE3 and a gRNA in the form of a preassembled ribonucleoproteinviacationic liposomes, serine-33 in β-catenin was edited to phenylalanine (TCT codon edited to TTT), allowing for the transdifferentiation of supporting cells into hair cells. Base editing of cochlear tissue was confirmedviasequencing,showing an editing rate between 0.7% and 3.0%depending on the region of the cochlea. In contrast,standard Cas9 editingviaHDR showed negligible signs of efficacy in cochlear cells. In subsequent work,Villigeret alused a variant of SaBE3, delivered into the liverviaadeno-associated viral (AAV) vectors, to directly correct a pathogenic T→C mutation in thePahgene with an editing rate as high as 29% and thereby treat the disease phenylketonuria in adult mice[26].
Adenine base editing has also been demonstrated to generate mutations in mice. Ryuet alused ABE7.10 to correct a nonsense mutation causing Duchenne muscular dystrophy inDmdmutant mice[27]. In this study, a dual trans-splicing AAV strategy was used to deliver ABE7.10 into the muscle. Intramuscular injection repaired the nonsense mutation with a 3.3%correction rate, restoring dystrophin production in around 17% of myofibers. In a more recent study, Wuet aldelivered ABE6.3viatail-vein hydrodynamic injection into mice to correct a point mutation that causes exon skipping in the fumarylacetoacetate hydrolase (Fah) gene, thereby ameliorating a mouse model for hereditary tyrosinemia type 1[28]. An editing efficiency of around 9.5% was observed in hepatocytes 32 days after treatment; no significant indel rate was detected. Codon-optimized ABE6.3(RA6.3) was delivered in lipid nanoparticlesviathe tail vein in a separate experiment; editing rates of around 0.4% were observed[28].
The use of base editors to correct deleterious mutations in human embryos has been explored.Lianget alused BE3 to correctHBB-28, an A→G mutation causing β-thalassemia[29]. By injecting BE3 mRNA and gRNA directly into embryos, they were able to achieve substantial base editing efficiencies at the targeted region. In 9 out of 22 tested embryos, a G→A correction was observed forHBB-28; in a single embryo, a G→C edit was observed instead.
Practical example—"reversible" base editing
To demonstrate the utility of the various base editors that are now available, we used both a cytosine base editor and an adenine base editor to explore the feasibility of treating patients with the disease hereditary tyrosinemia type 1 (HT1) caused by a specific founder mutation. HT1 is an autosomal recessive disorder caused by a deficiency of FAH,resulting in the inability of hepatocytes to properly catabolize the amino acid tyrosine and in the accumulation of toxic intermediate metabolites[30]. In the absence of treatment, HT1 can result in fulminant liver failure and death soon after birth. TheFAHc.1062+5G>A splice site mutation has an estimated carrier rate as high as 1 in 20 people in the Saguenay-Lac-Saint-Jean region of Québec, Canada, and due to the fact that more than 1 in 2000 newborns in that region had the disease, universal newborn screening for HT1 was instituted in Québec in 1970[31].
Because of the potential to directly correct the pathogenic G→A splice site mutation with an adenine base editor, we sought to test the efficacy of base editing against the mutant sequence. However, no human-derived cells with theFAHc.1062+5G>A splice site mutation were available. We noted that cytosine base editing with BE3 using a gRNA oriented towards a nearby PAM placed the site of the mutation within BE3's editing window, at position 5 in the protospacer sequence (Fig. 3). Thus, we first sought to use BE3 to introduce a C→T edit on the opposite strand (resulting in the precise G→A splice site mutation on the coding strand) in HEK 293T cells.
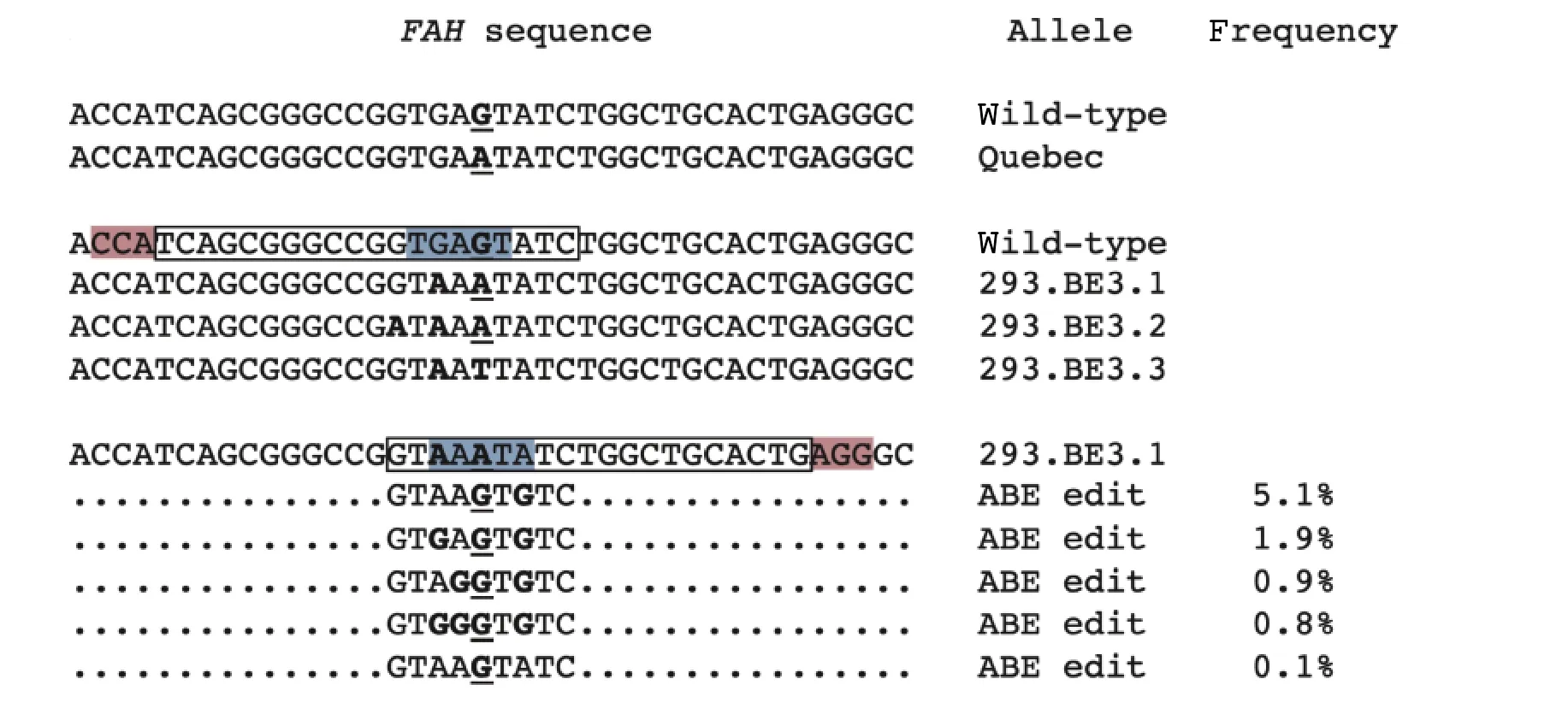
Fig. 3 Reversible base editing of a prevalent mutation responsible for hereditary tyrosinemia type 1 (HT1). The wild-type and"Quebec" alleles (FAH c.1062+5G>A splice site mutation) differ by a G→A change (bold and underlined). Cytosine base editing with BE3(protospacer in box, PAM in pink, editing window in blue) generated three different alleles in a HEK 293T clonal cell line, two alleles of which have the Quebec G→A mutation (bold and underlined; other changes are only in bold), designated 293.BE3.1 and 293.BE3.2.Adenine base editing with ABE7.10 (protospacer in box, PAM in pink, editing window in blue) and a gRNA perfectly matched to the 293.BE3.1 allele resulted in ≈9% of the 293.BE3.1 alleles in the treated cells having reversion of the Quebec G→A mutation to the wild-type G.
We transfected plasmids encoding BE3 (pCMVBE3, a gift from David Liu, Addgene plasmid No.73021) and the appropriate gRNA (protospacer+PAM=5′-GATACTCACCGGCCCGCTGA-3′+5′-TGG-3′)into cells and clonally expanded single cells a week following transfection. Out of approximately 20 clonal cell lines, only one displayed base editing at the target site. We expanded this cell line and again performed single cell cloning to ensure a "clean" clonal cell line.Upon deep sequencing of the target site in the resulting cell line, we observed that there were three distinct alleles (indicating that the cells were triploid for the chromosomal region in question, not surprising given the cells' immortalized state), none of them wild-type (Fig. 3). In one allele (293.BE3.1), the desired c.1062+5G>A mutation was present, along with an additional upstream G→A edit (position 7 in the protospacer sequence). In the second allele(293.BE3.2), the desired c.1062+5G>A mutation was present, along with two additional upstream G→A edits (positions 7 and 9 in the protospacer sequence).In the third allele (293.BE3.3), the site of the desired mutation instead harbored a G→T edit, along with one additional upstream G→A edit (position 7 in the protospacer sequence). Of note, the additional upstream G→A edits were at positions lying within or close to BE3's predicted editing window.
This exercise confirmed two known shortcomings of base editing. First, all of the potentially editable bases within the base editor's window can be edited,which can make it prohibitive to obtain alleles with just the desired base edit. This occurred in this example, where none of the alleles had only the desired c.1062+5G>A mutation. Second, the edits might not always have the expected outcomes. In this example, one of the alleles had a noncanonical G→T edit rather the desired (and likeliest) G→A edit.
Given the difficulty in obtaining a cell line with just the desired c.1062+5G>A mutation, we proceeded to use the triple-edited-allele cell line to assess whether adenine base editing of the mutation with ABE7.10 was feasible. We designed a gRNA oriented towards a nearby PAM whose protospacer matched only the first of the above-described three edited alleles(c.1062+5G>A mutation and one additional upstream G→A edit; designated 293.BE3.1), which should have the effect of constraining the activity of ABE7.10 to just that one allele and sparing the other alleles. The PAM placed the site of the c.1062+5G>A mutation within ABE7.10's editing window, at position 5 in the protospacer sequence (Fig. 3).
We transfected plasmids encoding ABE7.10(pCMV-ABE7.10, a gift from David Liu, Addgene plasmid No. 102919) and the appropriate gRNA(protospacer+PAM=5′-GTAAATATCTGGCTGCACTG-3′+5′-AGG-3′; deviation from wild-type sequence in bold and underlined) into the cell line. One week following transfection, we harvested genomic DNA from the cells and performed deep sequencing of the target site. Our previously described algorithm[12]identified the sequence reads from the first of the above-described three edited alleles (293.BE3.1) and discarded the other reads. The algorithm then determined the proportion of the selected reads in which the c.1062+5G>A mutation had been successfully corrected. We found that about 9% of the reads were corrected at that specific position (Fig. 3).However, we found that in almost all of the corrected reads, additional adenines had been converted to guanine (every possible combination of positions 3, 4,and 7 in the protospacer sequence). In a small proportion of the corrected reads only the c.1062+5G>A mutation was edited to guanine, again highlighting the challenging of restricting the editing activity of a base editor to just the desired position. Of note, we did observe that none of the reads harbored indel mutations.
Despite the inherent challenges, we were able to use a cytosine base editor and an adenine base editor to serially install and then correct a real-life pathogenic mutation in human cells. To our knowledge, this is one of the first reported demonstrations of "reversible"base editing. We note that HT1 is a disease in which any successfully corrected hepatocytes will have a selective advantage over uncorrected cells, and even a small proportion of corrected hepatocytes can ultimately reconstitute the entire liver and potentially cure the disease[24]. Thus, the obstacle presented by undesired additional A→G edits in the vicinity of a corrected c.1062+5G>A mutation is not insuperable from the perspective of therapeutic base editing for HT1. Nonetheless, as noted earlier, alteration of the Cas9 component of a base editor can serve to narrow the width of the editing window, and a suitably tailored variant of ABE7.10 might therefore achieve highly specific correction of only the c.1062+5G>A mutation.
Conclusions
Although the "CRISPR craze" that has taken the biomedical community by storm since early 2013 has captured the attention of not just scientists but the lay public, in fact it is base editing that might ultimately prove to be the truly revolutionary technology, as it permits precision editing in a manner that goes far beyond the capacity of standard CRISPR-Cas9 and previous generations of genome-editing tools, all of which have relied on DSBs and the difficult-toharness consequences of DSB repair. The ability to introduce or correct specific mutations—or both, which could permit reversible editing—with reasonably high efficiency, as demonstrated in the first wave of papers on base editing described here, as well as in the practical example presented here, portends a new therapeutic approach that might ultimately surpass standard genome editing. The coming years will undoubtedly see the development and refinement of base-editing tools as well as new approaches such as the recently described "prime editing"[32]that will open up the entire genome to precision editing.
Acknowledgments
This work was supported by U.S. National Institutes of Health grant R35-HL145203 and the Winkelman Family Fund in Cardiovascular Innovation (K.M.). K.M. is a co-founder and advisor of Verve Therapeutics and an advisor of Variant Bio.
 THE JOURNAL OF BIOMEDICAL RESEARCH2021年2期
THE JOURNAL OF BIOMEDICAL RESEARCH2021年2期
- THE JOURNAL OF BIOMEDICAL RESEARCH的其它文章
- Generating a CRISPR knockout mouse through a strong premature termination codon: a cautionary tale
- A transgenic pig model expressing a CMV-ZsGreen1 reporter across an extensive array of tissues
- Progress and challenges in CRISPR-mediated therapeutic genome editing for monogenic diseases
- Genome engineering technologies in rabbits
- Therapeutic gene editing strategies using CRISPR-Cas9 for the β-hemoglobinopathies
- Harnessing CRISPR-Cas system diversity for gene editing technologies