
Generating a CRISPR knockout mouse through a strong premature termination codon: a cautionary tale
2021-04-09 12:47:44QingRexLyu,PengYao,JosephM.Miano
Received: 11 July 2020; Revised: 09 November 2020; Accepted:16 November 2020; Published online: 25 December 2020
CLC number: Q789, Document code: A
The authors reported no conflict of interests.
This is an open access article under the Creative Commons Attribution (CC BY 4.0) license, which permits others to distribute, remix,adapt and build upon this work, for commercial use, provided the original work is properly cited.
Dear Editor,
Genetic inactivation or gene knockout (KO) has been a powerful method of elucidating gene function in mice. The traditional method of generating KO miceviatargeting mouse embryonic stem cells through homologous recombination is labor-intensive and of low efficiency. The emergence of clustered regularly interspaced short palindromic repeat(CRISPR) technology greatly simplifies the method of gene targeting at a reduced cost and with high efficiency. In its simplest form, CRISPR editing comprises two components, a single guide RNA(sgRNA) and Cas9 protein. Upon finding the complementary sequence preceding a protospacer adjacent motif (PAM), this ribonucleoprotein (RNP)complex generates a double-strand break (DSB) three nucleotides upstream of the PAM. The DSB is then repairedviaerror-prone non-homologous end-joining(NHEJ). Alternatively, a third component may be used involving a single-strand or double-strand repair template engineered with nucleotide substitutions,deletions or insertions of new genetic material[1]. The presence of such repair templates biases the reparation of the DSBviahomology directed repair (HDR)processes.
Multiple CRISPR strategies can be applied to achieve KO mice that include (a) the introduction of insertion or deletions (indels) around the coding sequence of interest leading to a frameshift that results, in most cases, in gene inactivation; (b) deletion of the promoter and first exon with two sgRNAs; (c)deletion of a downstream exon encoding a critical functional domain with two sgRNAs; (d) insertion ofloxP sites for conditional inactivation of a gene; and(e) insertion of a premature termination codon (PTC)downstream of the AUG codon[2]. In this study, we elected to use a single-strand repair template engineered to carry a strong PTC to generate a KO mouse translational arrest and subsequent nonsensemediated mRNA decay (NMD) of the truncated transcript.
KN motif and ankyrin repeat domains 1 (Kank1) is a cytoskeletal-associated protein that facilitates crosstalk between microtubules and integrin-based adhesions of the extracellular matrix[3]. We found Kank1 to be induced by the myocardin coactivator of smooth muscle contractile phenotype (GSE77120)[4].While a previous study implied its role in vascular development, no publication exists regarding a direct function of Kank1 in smooth muscle and, importantly,noKank1KO mouse has been reported. Our first objective was to validate expression ofKank1mRNA in adult mouse tissues. We found that Kank1 was expressed abundantly in smooth muscle-enriched tissues, including the aorta and bladder, consistent with GTEx portal data of humanKANK1mRNA expression (Supplementary Fig. 1AandB, available online).
A three-component CRISPR strategy was designed with a sgRNA targeting exon 5 ofKank1, Cas9 protein, and a single strand oligonucleotide repair template engineered with a strong PTC (Fig. 1A). The TAA nonsense codon has been shown to exhibit the highest activity in arresting protein translation[2]. To validate the efficiency of sgRNA, we cloned the predicted sgRNA target site with flanking 150-bp by PCR to perform anin vitroCas9 cleavage assay. The results revealed that the sgRNA effectively cleaved its target DNA segment (Supplementary Fig. 2, available online). Next,Kank1-PTC mice were generated by coinjecting sgRNA/Cas9 RNP and repair template with the PTC into mouse zygotes. Genotyping of founder mice revealed 5 out of 13 founders (38.4%) that carried the edited allele (Fig. 1B). TA cloning was then performed using the PCR products from positive founders. Three clones from each founder were picked for Sanger sequencing. The results verified that one founder carried the inserted TAATAAA at the edited site (Fig. 1CandD). This founder was then bred for germline transmission of the targeted allele.
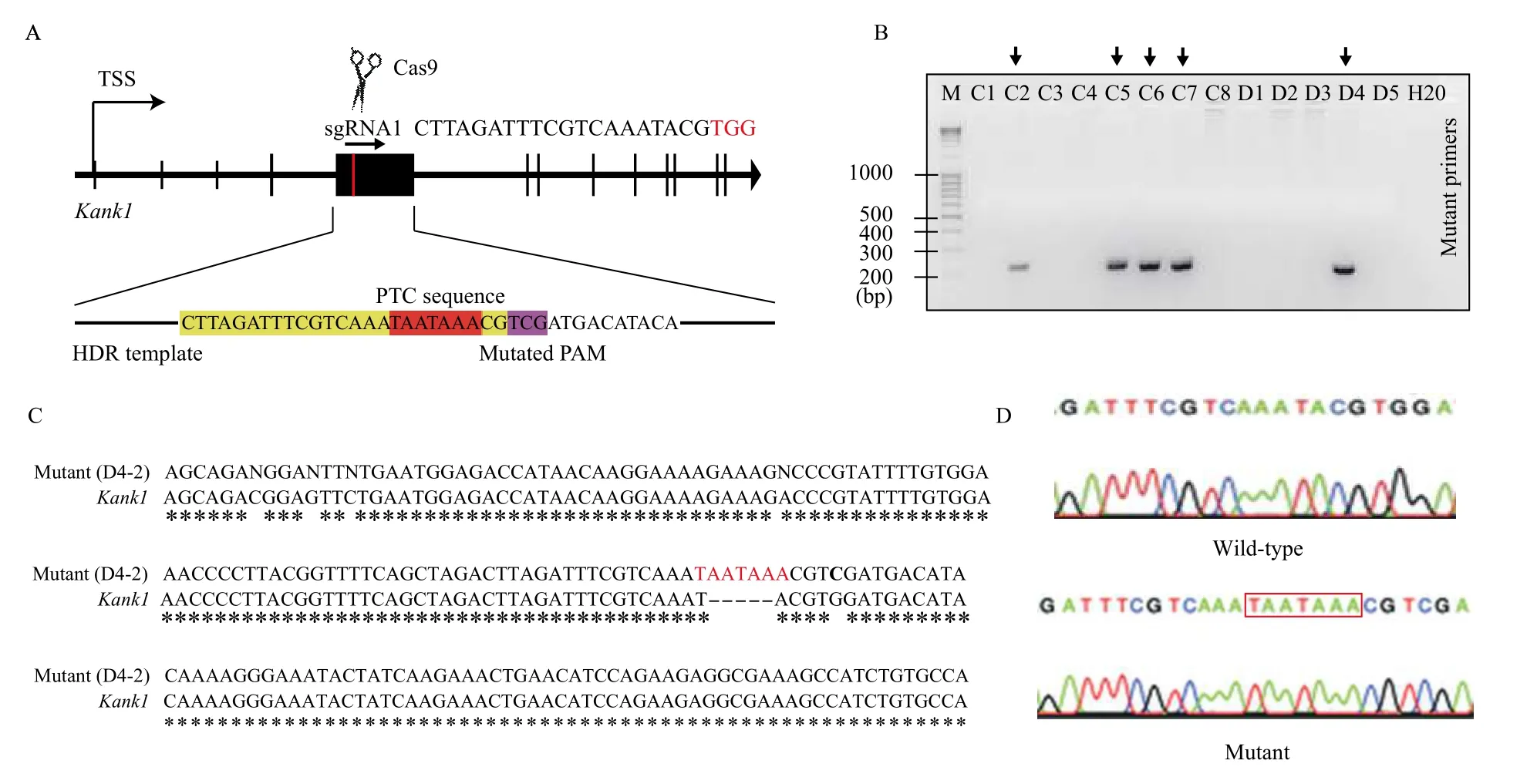
Fig. 1 Generating Kank1-PTC mouse. A: Strategy for generating Kank1-PTC mouse by inserting premature termination codon (PTC)downstream of first AUG start codon. The sgRNA1 and PAM (in red) sequence was shown. For the HDR template, the nucleotides highlighted in red are PTC sequence, the nucleotides highlighted in yellow are sgRNA sequence, nucleotides highlighted in pink are mutated PAM sequence. B: The genotyping agarose gel running image indicates five positive edited clones. C: The Sanger sequencing of a selected clone from gene edited pool, the clone D4-2 carries inserted PTC sequence TAATAAA, and the PAM is mutated from TGG to TCG. D: The chromatography of Sanger sequencing result for comparing wild-type (WT) and edited (Kank1-PTC) mouse.
HomozygousKank1-PTC mice were indistinguishable from their wild-type littermate controls (not shown). To examine whether levels of Kank1 were reduced by the PTC, we dissected the aorta and bladder from homozygousKank1-PTC mice (strain C57BL/6J) and wild-type littermate controls. Total RNA was harvested and real-time qPCR results revealed little to no change inKank1mRNA in both aorta and bladder (Fig. 2A). To determine if changes were evident in Kank1 protein levels, Western blotting was performed in these tissues. Similar to results observed with real-time qPCR, we found no evidence of loss of Kank1 protein in the aorta and bladder(Fig. 2B). These expression data indicate that our strategy to inactivate theKank1gene through precision targeting of a strong PTC was unsuccessful.
To generate aKank1KO mouse in a more aggressive manner, we performed 2-component CRISPR genome editing[3]using Cas9 protein and sgRNAs flanking the largest coding exon (exon 5) ofKank1(Fig. 2C). PCR genotyping revealed 1 out of 19 founder mice (5.2%) with successful deletion of exon 5 (data not shown). DNA alignment of Sanger sequenced mutant DNA showed precise deletion of 2970 bp DNA segment spanningKank1exon 5(Fig. 2D). There was no obvious phenotype observed in this founder mouse. We crossed the founder mouse with wild type C57BL/6J mice and continuously bred the mice to generate 4 litters, yielding a total of 32 pups. None of the pups genotyped positive for the expected deletion (data not shown). Thus, we were unable to breed this deletion through the germline.Accordingly, we collected the aorta and bladder from the single founder mouse for Western blotting. The results showed a reduction ofKank1protein in the aorta, but not in the bladder (Fig. 2E). These findings are consistent with the fact that founder mice derived from CRISPR editing are mosaic and would not be expected to carry the deleted allele in all cells of the adult mouse.
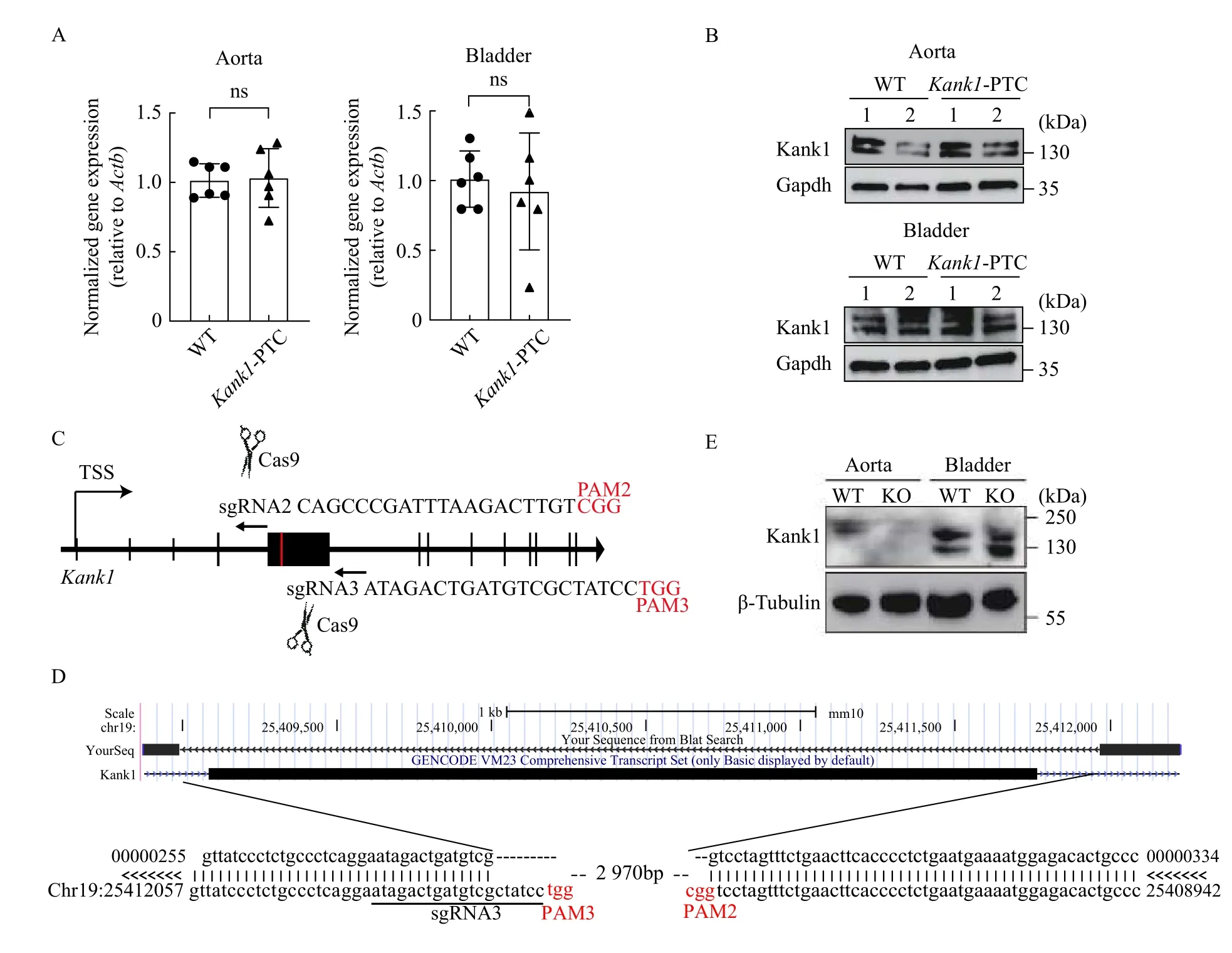
Fig. 2 Validation of Kank1 protein level in gene edited mice. A: Real-time qPCR results for wild-type (WT) and gene knockout (KO)mice (Kank1-PTC) using mouse aorta (n=6, ns indicates no statistical significance). B: Western blotting using anti-KANK1 antibody with aorta and bladder from WT vs. Kank1-PTC mice. C: The schematics for generating Kank1 KO mouse by deleting the exon 5 with two sgRNAs. D: The Sanger sequencing alignment results for the Kank1 KO mice, a DNA fragment (2970 bp) has been removed from chromosome 19 by sgRNA2 and sgRNA3 (PAM sequences are labeled in red). E: Western blotting using anti-KANK1 antibody with aorta and bladder from WT vs. Kank1 KO mice.
In this study, we intended to generate aKank1KO mouse by inserting a strong PTC downstream of the initiating AUG codon. However, expression studies failed to reveal loss of Kank1 protein in the edited mice. To discuss the underlying reason for this unexpected result, we re-analyzed the genomic sequence ofKank1and found there is an in-frame AUG start codon located 329 nucleotides downstream of our insertion site (Supplementary Fig. 3, available online). Therefore, although infrequently reported[5], it is possible that the ribosome skipped the PTC and reinitiated translation at the second AUG codon producing a minor truncated KANK1 protein.Alternatively, the post-termination ribosomes could reinitiate translation at downstream AUG codons,which allows the transcript to evade the NMD pathway[6]. Due to the size of the Kank1 protein(approximately 140 kDa) and the fact that the Nterminal truncated protein carrying the PTC comprises only 47 amino acids, we could not distinguish them with Western blotting, and we used the Kank1 antibody to recognizeKank1C-terminal(Supplementary Table 2, available online). Surprisingly, the humanKANK1gene possesses more than 20 different variants, whereas the database from UCSC Genome Browser only showed 2Kank1variants in mice. Thus, we considered that there could be a similar number of un-annotated mouseKank1variants due to the understudy of mouseKank1. Thus,our original editing strategy could place a premature stop codon downstream of one initiator methionine start codon but upstream of a second, which could conceivably result in the unsuccessful knockout ofKank1. In addition, we also noticed that the molecular weight of Kank1 in the aorta appears larger than in the bladder (Fig. 2E). This discrepancy is unexplored at this time but one of the possibilities is that Kank1 in the aorta underwent posttranslational modification which confers the protein with slower migration velocity. This interpretation needs to be investigated and verified in future work.
There are many strategies to generate KO mice using CRISPR-Cas9 genome editing. Two-component CRISPR can be applied to create a DSB at the target site and NHEJ repairs DNA in an error-prone manner,which will create indels at the editing site and a frameshifted mRNA transcript. However, there is a 1/3 possibility to obtain the indel with in-frame editing, which could have no detectable change in protein expression of the target gene. It should be noted that even in the presence of a frame-shifted transcript with extinguished protein expression, the edited mRNA transcript could exert a noncoding function if it is not targeted for NMD. Similarly, the internal exon deletion strategy using two sgRNAs may also result in a truncated mRNA with unknown function. A popular strategy is to remove the promoter and first exon of the target gene with two sgRNAs.However, there is increasing evidence for antisense transcription of long noncoding RNAs whose deletion could confound interpretation. The same can be said for the excision of regulatory elements controlling a distal gene. This approach may also inactivate the expression of intron-embedded small RNAs (snoRNA,miRNA, circular RNA,etc.) in the sense strand premRNA. Moreover, targeting a promoter and first exon may in some instances result in the loss of only one isoform of the gene when internal promoter-driven isoforms exist. The PTC insertion model is an ideal approach to inactivate a target gene with minimal disturbance of local sequences. The PTC insertion would be expected to trigger NMD leading to target mRNA degradation with little disturbance of neighboring genes.
NMD is a well-studied cellular process for mRNA quality control. There are two different mechanisms for NMD: 3′-UTR exon-junction complex (EJC)-dependent NMD, and EJC-independent NMD[7]. In our study, the insertion of a PTC in exon 5 ofKank1was expected to initiate EJC-dependent NMD because the termination codon resides approximately 2600 nucleotides upstream of an exon-exon junction; this would fulfill the so-called "50-55 rule" for NMD[7].However, our results indicate that the ribosomal machinery somehow read through or skipped the PTC site and re-initiated translation from a downstream start codon resulting in a slightly truncated protein that we could not resolve given the high molecular weight of full-lengthKank1.
What are the underlying mechanisms for this failed targeted KO experiment? The reasons could be: a) the insertion of a PTC sequence creates an unusually long(>3 kb) 3′-UTR which compromises the translation termination effect, and for unknown reasons, NMD is not triggered; b) the ribosome may initiate translation at a downstream AUG start codonviabinding to a potential internal ribosome entry site or bypassing the first start codon during mRNA scanning. In this context, an analysis of 193 frameshifts following CRISPR-mediated gene deletions in 136 distinct gene loci demonstrated some level of protein expression in more than 30% of the cases, implying substantial gene hypomorphism[8]. Mechanisms for such residual protein expression included re-initiation of translation and exon skipping[8]. Interestingly, alternative inframe translation initiation has been observed forCx43mRNA[9]. And c) the position of the PTC is in close proximity to the initiating AUG (47 amino acids downstream in the present case) that NMD is rendered inefficient as shown in an initial report and discussed in a subsequent review[10].
The precise mechanism(s) for sustained Kank1 protein expression with the introduction of a strong PTC is unknown. However, the results shown here underscore the importance of careful deliberation when contemplating CRISPR-mediated gene KO and,in particular, the targeted insertion of a PTC to arrest translation.
We thank the University of Rochester Genome Editing Core for performing the microinjection experiments. Our work was supported by the National Institutes of Health under grant No. HL-138987 to JMM and American Heart Association Postdoctoral Fellowship (No. 17POST33660938 to QRL).
Yours Sincerely,
Qing Rex Lyu1,2,3,?, Peng Yao1, Joseph M. Miano1,21Aab Cardiovascular Research Institute,
University of Rochester School of Medicine &Dentistry,
Rochester, NY 14642,
USA;
2Vascular Biology Center,
Medical College of Georgia at Augusta University,
Augusta, GA 30912,
USA;
3Institute of Biomedicine and Health,
Chongqing Institute of Green and Intelligent Technology,
Chinese Academy of Sciences,
Chongqing 400714,
China.
?Tel: +86-18575532263, E-mail: lvqing@cigit.ac.cn.
 THE JOURNAL OF BIOMEDICAL RESEARCH2021年2期
THE JOURNAL OF BIOMEDICAL RESEARCH2021年2期
- THE JOURNAL OF BIOMEDICAL RESEARCH的其它文章
- A transgenic pig model expressing a CMV-ZsGreen1 reporter across an extensive array of tissues
- Progress and challenges in CRISPR-mediated therapeutic genome editing for monogenic diseases
- Genome engineering technologies in rabbits
- Therapeutic gene editing strategies using CRISPR-Cas9 for the β-hemoglobinopathies
- Base editing: a brief review and a practical example
- Harnessing CRISPR-Cas system diversity for gene editing technologies