
Investigation of the interaction between indigotin and two serum albumins by spectroscopic approaches
2013-12-23 06:14ZhengJunChengHongMeiZhoQinYongXuRongLiu
Zheng-Jun Cheng, Hong-Mei Zho, Qin-Yong Xu, Rong Liu
aChemical Synthesis and Pollution Control Key Laboratory of Sichuan Province, China West Normal University,Nanchong 637009, China
bNanchong Petrochemical School, Nanchong 637000, China
1. Introduction
Indigotin (Fig.1) is one of the oldest natural dyes, which has been obtained from a wide variety of plants such as Indigofera spp. (Africa, Asia and South America), Polygonum tinctorium(China and Korea) and Isatis tinctoria (Europe) [1]. It has been widely used as colorant in common foods such as sweets,drinks and ice cream.Like many other food additives,control the dosage of indigotin is of considerable importance in food industry because of its toxic and carcinogenic potential [2].It was reported that some dyes would stain certain tissues;however, these dyes would selectively stain, combine with and destroy pathogenic organisms without causing appreciable harm to the host[3].Nowadays,much research on the binding of drugs to proteins has been carried out [4-11], but seldom report on the binding interactions of proteins with dyes. The study of the interaction between indigotin and proteins was not only important to comprehend its transport and metabolism process in the body, but also it could help us know the toxicity of indigotin at the functional macromolecular level,especially at lower concentrations.

Fig.1 Molecular structures of indigotin.
Serum albumins (SAs) are the major soluble protein constituents of the circulatory system, and constitute 50-60% of total amount of plasma proteins [12]. The most important property of this group of proteins is that they serve as transporters for a variety of compounds such as drugs,fatty acids and amine-terminated dendrimers [13]. In this work,bovine serum albumin (BSA) and human serum albumin(HSA) were selected as the models for evaluating the interactions of dyes with proteins because of their similar folding and well-known primary structure [11]. From the spectroscopic point of view, one of the main differences between the two proteins is that HSA has only one tryptophan residue (Trp-214) in sub-domain IIA, whereas BSA has two tryptophan residues (Trp-134 and Trp-212), that is to say, Trp-134 in the first domain is located on the surface of the molecule,and Trp-212 in the second domain is located within a hydrophobic binding pocket of BSA [14]. Because investigation on the interactions between dyes and proteins can help us better understand the absorption and distribution of dyes, it would become an important research field in chemistry, life sciences and clinical medicine.
In the paper, the binding interactions of indigotin to BSA/HSA have been investigated by various spectroscopic methods. The binding constants, the thermodynamic parameters,the number of binding sites,the binding forces and the energy transfer distance of indigotin-BSA/HSA complexes were estimated. At the same time, the effect of indigotin on the microenvironment and conformation of proteins was also discussed. In addition, the binding mechanism of indigotin with proteins was analyzed,and the effects of common ions on the binding constants of indigotin-BSA/HSA complexes were explored.
2. Materials and methods
2.1. Reagents and chemicals
Human serum albumin (HSA, 96% purity) acquired from Sigma chemical company (St. Louis,USA), and bovine serum albumin (BSA, Fraction V, 98% purity) purchased from Roche Company, were used without further purification,and they were both dissolved in the Tris-HCl buffer solution(pH 7.4)to form a solution of 2.0×10-4M and then stored in the dark at 4°C; Indigotin (98% purity) was purchased from Aladdin Chemistry Co., Ltd. (Shanghai, China) and its stock solution was prepared by dissolving it in doubly distilled water with the final concentration of 6.0×10-4M; Phenylbutazone(99+% purity) was purchased from ACROS ORGANICS(New Jersey, USA); Ibuprofen (99% purity) was purchased from Shanghai Civi Chemical Technology Co., Ltd. (Shanghai, China); Digitoxin (99% purity) was purchased from Hubei Huabei Biomedicine Co., Ltd. (Wuhan, China). All other materials were of analytical reagent grade and doubly distilled water was used throughout the experiment.
2.2. Spectroscopic measurements
All fluorescence spectra were carried out on a Cary Eclipse fluorescence spectrofluorometer (Varian, USA) equipped with a xenon lamp source and 1.0 cm quartz cell and a thermostat bath. 3.0 mL of BSA/HSA solution was added accurately into the quartz cell and then titrated by the successive additions of indigotin solution with the concentration of 6.0×10-4M using a 50 μL microsyringe to attain a series of final concentrations. Titrations operated manually and mixed moderately.The fluorescence spectra were recorded in the range of 250-500 nm (excitation wavelength 280 nm) using 5/5 nm slit widths at different temperatures (293, 298, 304 and 310 K).Synchronous fluorescence spectra were recorded from 220 to 320 nm at Δλ=15 and Δλ=60 nm, respectively. In addition,the fluorescence spectra of indigotin-BSA/HSA systems were also determined in the presence of some common ions(such as Ca2+,Zn2+,Mn2+,Fe3+,Cu2+,Cl-,CH3COO-and CO32-)in the range of 250-500 nm at 298 K. Also appropriate blanks corresponding to the buffer were subtracted to correct the fluorescence background.
The competitive experiments between indigotin and three site markers (ibuprofen, phenylbutazone and digitoxin) for BSA/HSA were performed using the fluorescence titration method.The concentrations of BSA,HSA and the site marker were all stabilized at 5.0×10-6M. The solution of indigotin was gradually added into the BSA-ibuprofen, BSA-phenylbutazone, BSA-digitoxin, HSA-ibuprofen, HSA-phenylbutazone,or HSA-digitoxin mixtures. The fluorescence quenching data for six systems were recorded in the range of 250-500 nm using an excitation wavelength of 280 nm.
UV-vis absorption spectra were collected on a UV-3600 spectrophotometer (Shimadzu, Japan) equipped with 1.0 cm quartz cells at 298 K.And UV-vis absorption spectra of BSA/HSA in the presence and absence indigotin were recorded in the range of 230-500 nm.The concentration of BSA/HSA was fixed at 5.0 μM while those of indigotin changed from 0 to 10.0 μM.
FT-IR measurements were carried out on a Nicolet Nexus 6700 FT-IR spectrometer (America) via the attenuated total reflection(ATR)at a resolution of 4 cm-1and 64 scans.FT-IR spectra of HSA(0.10 mM)and BSA(0.10 mM)in the presence and absence of indigotin were recorded in the range of 400-4000 cm-1at 298 K. The molar ratio of dye/protein was maintained at 3:1. The corresponding absorbance contributions of the buffer and indigotin solutions were recorded and digitally subtracted with the same instrumental parameters.
Three-dimensional fluorescence spectra were performed under the following conditions: the initial excitation wavelength was set at 200 nm with increment of 2 nm, the number of scanning curves was 76; the emission wavelength was recorded between 200 and 500 nm at a scanning rate of 24000 nm/min, and other scanning parameters were the same as those of the fluorescence quenching spectra.
CD spectra of BSA/HSA in the absence and presence of indigotin were performed on a Jasco-810 automatic recording spectropolarimeter (Japan) using a 0.1 cm cell at room temperature. All the CD spectra were recorded in the range of 200-260 nm at a scanning rate 100 nm/min. Spectra were corrected for buffer absorbance and recorded at the molar ratio of proteins/dye of 1:10 and 1:20.
3. Results and discussion
3.1. The binding mechanism analysis of indigotin and HSA/BSA
Fluorescence spectroscopy has been widely applied to investigate the interactions of drugs to proteins,and hence it can be used to analyze the quenching mechanism of proteins-drugs complexes. The fluorescence spectra of BSA/HSA in the presence of indigotin are shown in Fig.2. As shown in Fig.2, the fluorescence intensities of BSA/HSA decreased with increasing the concentrations of indigotin, indicating the strong interactions between indigotin and BSA/HSA occurred[15]. From latter UV-vis absorption, fluorescence, FT-IR, 3D fluorescence and CD spectra, we could conclude that the indigotin-BSA and indigotin-HSA complexes were formed and the binding interactions affected the conformation of proteins.
Fluorescence quenching can proceed by different mechanisms, which are usually classified as either dynamic quenching or static quenching. For the dynamic quenching, as the temperature of the system rises, the effective collision time between molecules, the energy transfer efficiency and the fluorescence quenching constants of substances will all increase [16]. Whereas, for the static quenching, increasing temperature reduces the stability of complexes formed, resulting in the quenching constants decrease. In order to examine the quenching mechanism, we first assumed that the fluorescence quenching was a dynamic quenching, thus the fluorescence quenching data were analyzed by the Stern-Volmer equation [17]:
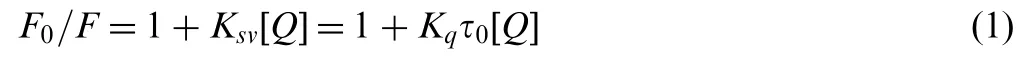
where F0and F denote the steady-state fluorescence intensities in the absence and presence of quencher, respectively. Ksv,Kq,τ0and [Q] are the Stern-Volmer quenching constant, the quenching rate constant of biomolecule,the average lifetime of protein without the quencher, and the concentration of quencher, respectively. The values of Ksvcould be calculated by linear regression of a plot of F0/F versus [Q]. Obviously,

Taking the fluorescence lifetime of the biopolymer (τ0) as around 10-8s [16], the values of Kqcould be obtained according to Eq. (2).
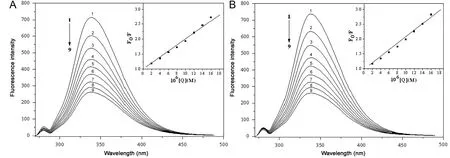
Fig.2 Fluorescence spectra of BSA(A)and HSA(B)in the presence of various concentrations of indigotin,c(BSA)=c(HSA)=5.0 μM;c(indigotin) (μM), 1-9: 0, 2.0, 4.0, 6.0, 8.0, 10.0, 12.0, 14.0 and 16.0, respectively. The insets correspond to the Stern-Volmer plots.T=298 K.
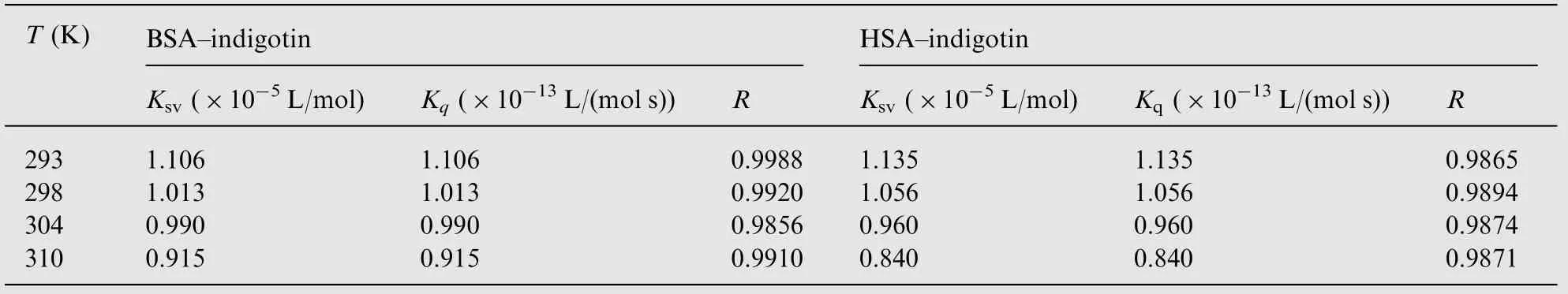
Table 1 The parameters of Stern-Volmer plots for the indigotin-BSA/HSA systems at different temperatures.
The insets of Fig.2 display the Stern-Volmer plots for the quenching of BSA/HSA by indigotin at 298 K. The values of Ksv, Kqand R at four different temperatures (293, 298, 304 and 310 K) are listed in Table 1. As shown in Table 1, the values of Ksvdecreased with increasing temperature. Moreover, the values of Kqwere far greater than the maximum scattering collision quenching constant value of the biomolecule (2×1010L/(mol s)) [18]. The results indicated that the quenching mechanism of indigotin-BSA/HSA was initiated by the complex formation rather than the dynamic collision. In addition, the quenching data were further analyzed by the static quenching equation [19]:

In the present case, (F0-F) is the difference in fluorescence intensities between the absence and presence of quenching compound at the concentration [Q], Kais the effective quenching constant for the accessible fluorophores, and fais the fraction of accessible fluorophore to a polar quencher, which indicates the fractional fluorescence contribution of the total emission for an interaction with a hydrophobic quencher. The plots of F0/(F0-F)versus 1/[Q] yield 1/faas the intercept, and 1/faKaas the slope(Fig.3). Thus, the ratio of the intercept and the slope gives the value of Ka. The corresponding Kavalues for the indigotin-BSA complex were obtained to be 9.579×104L/mol, at 293 K;8.731×104L/mol, at 298 K; 7.613×104L/mol, at 304 K;5.204×104L/mol, at 310 K. The values of Kafor the indigotin-HSA complex were obtained to be 7.645×104L/mol, at 293 K;5.842×104L/mol, at 298 K; 5.117×104L/mol, at 304 K;2.886×104L/mol,at 310 K.The decreasing trend of the Kavalues with increasing temperature was in accordance with the dependence Ksvvalues on temperature as mentioned above.Therefore, the result reconfirmed that the quenching mechanism between indigotin and BSA/HSA belonged to the static quenching.
Besides, fluorescence quenching mechanism can be also confirmed by the analysis of Stern-Volmer plots at different concentrations of proteins. The value of Ksvis expected to depend on the concentration of BSA/HSA in a static quenching process, and yet its value does not change at any concentration of BSA/HSA in only a dynamic process [20]. The plots of F0/F versus [Q] yield Ksvas the slope (not show the figure). The corresponding values of Ksvand R for indigotin-BSA/HSA complexes are shown in Table 2. It could be seen from Table 2 that the values of Ksvdecreased with increasing the concentrations of proteins for two systems. The results suggested that the quenching mechanism of BSA-indigotin and HSA-indigotin systems was initiated by the static quenching.
3.2. UV-vis absorption spectra
UV-vis absorption measurement is a very simple and effective method,which is often applied to explore the structural changes of protein and investigate the ligand-protein complex formation.Indigotin (Fig.4) showed a strong absorption around 280 nm,overlapping with proteins.Upon addition of indigotin into serum albumins solution, the peak at 280 nm was gradually increased.Since both indigotin and proteins have absorption at 280 nm,it is unable to predict whether the increase in the absorption might be due to the interaction between the molecules or to increase in the concentration of indigotin.As a consequence of this fact,we have recorded UV absorption spectra of indigotin,free BSA/HSA and[indigotin-BSA/HSA complex]-[ indigotin] systems (Fig.4).As could be seen from Fig.4, the absorption intensities of BSA/HSA increased with the addition of indigotin and the maximum peak position of the indigotin-BSA/HSA systems red shifted(from 278 to 281 nm),which indicated that indigotin could bind to proteins and the indigotin-BSA/HSA complexes were formed and in the polarity around the tryptophan residues altered [21].
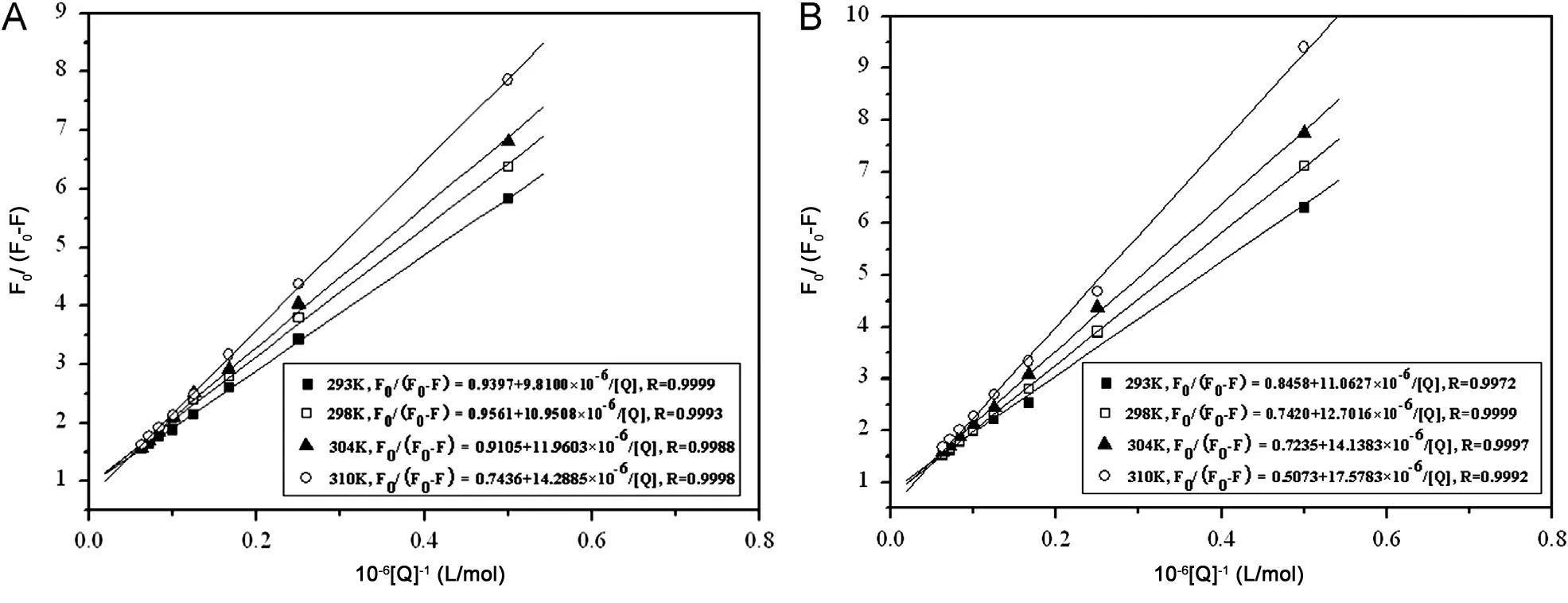
Fig.3 Modified Stern-Volmer plots for BSA (A) and HSA (B) with indigotin at (■) 293 K, (□) 298 K, (▲) 304 K and (O) 310 K.
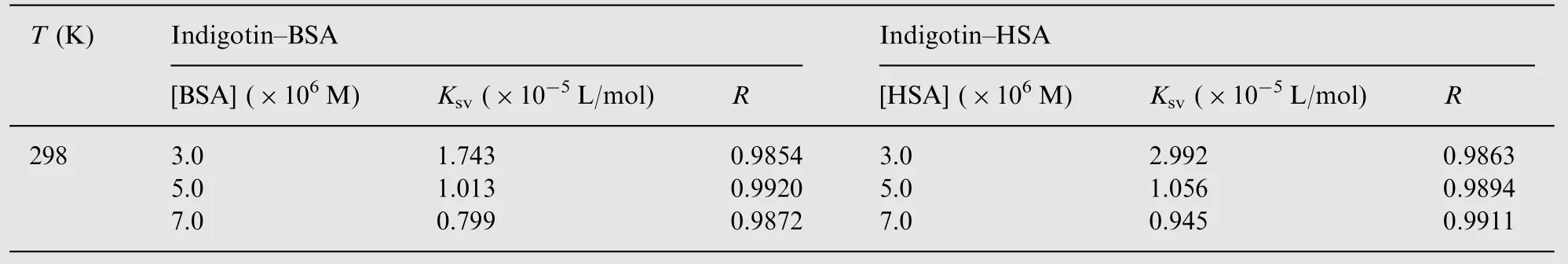
Table 2 The parameters of Stern-Volmer plots for the indigotin-BSA/HSA systems at different concentrations of proteins,T=298 K.

Fig.4 UV-vis absorbance spectra of [BSA-indigotin complex]-[indigotin] (A) and [HSA-indigotin complex]-[indigotin] (B); the concentrations of BSA and HSA were kept at 5.0 μM (a); the indigotin concentrations for two systems were maintained at 2.00 μM(b), 4.00 μM (c), 6.00 μM (d), 8.00 μM (e) and 10.00 μM (f); a concentration of 2.00 μM (g) was used for indigotin only.

Fig.5 Plots of log [(F0-F)/F] versus log [Q] for the BSA-indigotin (A) and HSA-indigotin (B) systems at four different temperatures,cBSA=cHSA=5.0 μM, pH=7.4.
3.3. The binding parameters and binding interaction force of BSA/HSA with indigotin
If we assume that there are independent binding sites in the biomolecule, the binding constant (K) and the number of binding sites (n) can be calculated using the equation shown below [22]:
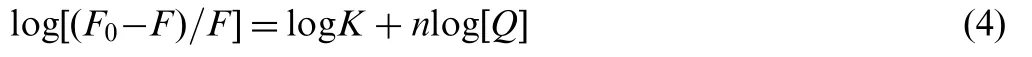
where F0, F, and [Q] are the same as in Eq. (1); n is the number of binding sites per albumin molecule; K is binding constant of indigotin with BSA/HSA. The binding constant(K)and the number of binding sites(n)could be calculated by plotting log[(F0-F)/F]versus log[Q]as shown in Fig.5.From the slope(n)and the intercept(log K)of the linear fitting plots,the binding constant K values are calculated and are listed in Table 3. As could be seen in Table 3, the binding constants calculated for indigotin-BSA/HSA systems suggested low binding affinity compared to other strong protein-ligand complexes with binding constants ranging from 106to108M-1[23,24].However,lower binding constants(103-104M-1)were also reported in several other ligand-protein complexes using fluorescence spectroscopic methods [25-27]. In addition, the binding constants (K) increased with increasing temperature,which indicated that the stability of the indigotin-BSA and indigotin-HSA complexes would decrease. The correlation coefficients (R) were larger than 0.9960, suggesting that assumptions underlying the derivation of Eq. (4) were reasonable. It could be inferred from the values of n that there was about one independent class of the binding site on BSA/HSA in the presence of indigotin. HSA has one tryptophan residue (Trp-214) in subdomain IIA, whereas BSA hastwo tryptophan residues (Trp-134 and Trp-212). Trp-134 residue is embedded in the subdomain IB and it is more exposed to a hydrophilic environment, and Trp-212 residue is embedded in subdomain IIA and it is deeply buried in the hydrophobic loop. Such, indigotin most likely bind to the hydrophobic pocket located in subdomain IIA [28]. Whereas,larger number of bound lipid (n) was reported for HSA with DDAB (n=1.82), DOTAP (n=1.56), and DOPE (n=1.76)[29].

Table 3 The binding constants K,the number of binding sites n,and the thermodynamic parameters of the indigotin-BSA/HSA systems at four temperatures.
The interactions between small molecules and macromolecules usually follow four binding forces: hydrogen bond,van der Waals,electrostatic and hydrophobic force.The binding studies were carried out at 293, 298, 304 and 310 K. At these temperatures, BSA and HSA do not undergo any structural change. In order to characterize the forces between indigotin and BSA/HSA, the thermodynamic parameters on four temperatures were analyzed by the Van't Hoff equation [30]:
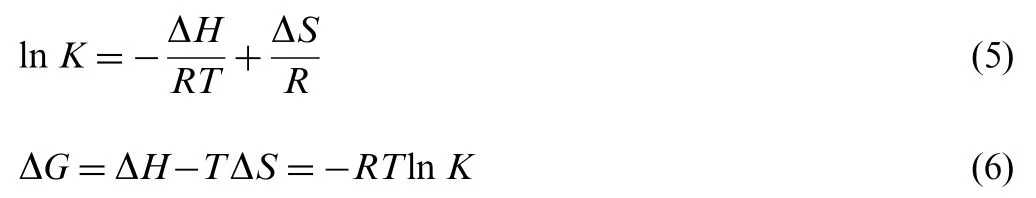
K is the equilibrium binding constant (L/mol), T is absolute temperature and R is the gas constant (8.314 J/(mol K)).According to the binding constants of indigotin-BSA/HSA systems obtained at the four temperatures mentioned earlier,the corresponding values of ΔH and ΔS can be obtained from the linear relationship between ln K and the reciprocal absolute temperature(Fig.6),and the ΔG values can be calculated by Eq.(6). The values of ΔH, ΔS and ΔG are summarized in Table 3.As shown in Table 3, the negative values of ΔG indicated that the binding interactions of indigotin to BSA/HSA were spontaneous processes.The positive ΔH and ΔS values suggested that the indigotin-BSA/HSA complexes were formed by the hydrophobic interaction [31].
3.4. Identification of binding sites on BSA/HSA
In order to further identify the binding sites on BSA/HSA by indigotin, the site marker competitive experiments are performed using the drugs specifically bind to a know site or region on BSA/HSA. The proteins (BSA and HSA) consist of three homologous domains (I-III), and each domain contains two subdomains (A and B). The binding sites of endogenous or exogenous ligands to BSA/HSA may be in these domains,and the principal regions of the binding sites of proteins are located in hydrophobic cavities in subdomains IIA or IIIA.Moreover, many ligands could bind specifically to HSA/BSA,for example,phenylbutazone and warfarin for site I,ibuprofen and flufenamic acid for site II,and digitoxin for site III[32].In the study, the site probes, viz. phenylbutazone, ibuprofen and digitoxin were chosen to identify the binding site on BSA/HSA by indigotin. The fluorescence spectra of the ternary mixtures(indigotin- proteins-the site probe) were recorded in the range of 250-500 nm. That is to say, the solution of indigotin was gradually titrated to the system of BSA/HSA with the site marker and the concentration of BSA/HSA to the site marker was kept at 1:1. Then, the fluorescence quenching data were analyzed using Eq. (4),and the values of K and R are listed in Table 4. As shown in Table 4, the binding constants of indigotin-BSA/HSA systems remarkably decreased in the presence of phenylbutazone (PB), whereas with the addition of ibuprofen (IB) or digitoxin (DIG) resulted in only a small difference. The results indicated that the binding sites of indigotin to BSA/HSA mainly located within site I (subdomain IIA), suggesting that the involvement of hydrophobic interaction played a major role in the binding reactionbetween indigotin and BSA/HSA.Obviously,the above results were in good agreement with those obtained by the binding parameters and binding interaction forces of BSA/HSA with indigotin studies.

Table 4 Effects of the site probe on the binding constants of indigotin to BSA/HSA.

Fig.7 The spectral overlap of UV-vis absorption spectrum of indigotin and fluorescence emission spectra of BSA(A)and HSA(B);(a)fluorescence emission spectrum of BSA; (b) UV-vis absorption spectrum of indigotin; and (c) fluorescence emission spectrum of HSA.c(BSA)=c(HSA)=5.0 μM, c(indigotin)=5.0 μM, T=298 K.
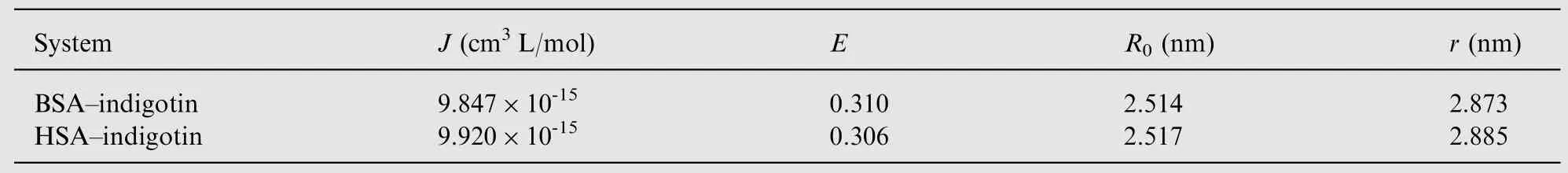
Table 5 The energy transfer parameters between indigotin and BSA/HSA.
3.5. Energy transfer between indigotin and BSA/HSA
The distance between BSA/HSA and indigotin can be calculated by Fster's non-radiative energy transfer theory [33].According to the theory, the efficiency of energy transfer is influenced by the distance between acceptor and donor, and the critical energy transfer distance is calculated using Eq. (7)and the value of R0is obtained by Eq. (8)

In the above experiment conditions, k2=2/3, n=1.36,Φ=0.15 for BSA [34] and k2=2/3, n=1.336, Φ=0.118 for HSA [35]. The value of J can be calculated by the following equation:
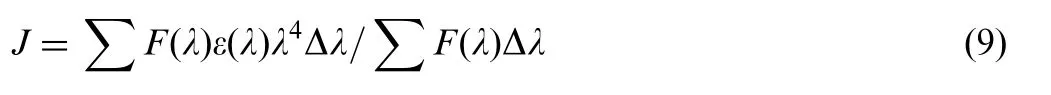
The overlap of UV-vis absorption spectrum of indigotin and fluorescence emission spectra of BSA and HSA are shown in Fig.7.
According to Eqs.(7-9)and the experimental data,the J,E,R0and r values of the binding interaction between indigotin and BSA/HSA are obtained and listed in Table 5.As shown in Table 5, the average distance of indigotin to BSA/HSA was shorter than 8 nm, suggesting that the non-radiative energy transfer from BSA/HSA to indigotin occurred with high probability and the result (R0<r) reconfirmed the presence of the static quenching interaction between indigotin and BSA/HSA [36]. Moreover, the binding distance of indigotin to proteins was shorter than those of ribavirin to BSA/HSA(5.43 nm/3.74 nm) [37], which indicated that the binding affinity of indigotin-proteins complexes was higher than that of ribavirin-BSA/HSA complexes. The result was consistent with their fluorescence spectra data studies [38,39].
3.6. The influence of indigotin on the conformation BSA/HSA
In order to explore the structural change of BSA/HSA in the presence of indigotin, synchronous fluorescence, threedimensional fluorescence, FT-IR and CD spectra of BSA/HSA before and after the addition of indigotin were analyzed,respectively.
3.6.1. Synchronous fluorescence spectroscopy studies
Synchronous fluorescence spectrum is a useful method to evaluate the conformational changes of proteins. The shift in position of emission maximum reflects the change of the polarity around the chromophore molecule, thus the microenvironment of amino acid residues(such as tryptophan,tyrosine,etc.)can be studied by measuring the possible shift in maximum emission wavelength. When the wavelength interval (Δλ) between the excitation and emission wavelength is stabilized at 15 and 60 nm,the synchronous fluorescence spectra offer the characteristics of tyrosine and tryptophan residues, respectively. The fluorescence spectra of BSA/HSA with various amounts of indigotin are recorded at Δλ=15 nm and Δλ=60 nm and are shown in Fig.8.
It was apparent from Fig.8(A, a) and (B, a) that the emission intensities of tyrosine residues decreased and the emission maximum wavelength slightly shifted towards short wavelength (from 286 to 284 nm), indicating that the interaction of indigotin with BSA/HSA affected the conformation of the region around tyrosine residues.In Fig.8(A,b)and(B,b),the emission maximum wavelength of tryptophan residues showed slight blue shifts (from 280 to 278 nm) with the addition of indigotin,which suggested that the hydrophobicity around tryptophan residues increased and the polarity around tryptophan residues decreased. The above results implied that indigotin was closer to tryptophan residues than tyrosine residues, namely the binding sites mainly were focused on tryptophan moiety.The result was consistent with the result of the displacement experiments.
3.6.2. Three-dimensional fluorescence spectra studies
Because three-dimensional fluorescence spectroscopy can provide more detailed information about the conformational changes of proteins, it has become a popular fluorescence analysis technique in recent years [40]. The excitation wavelength, emission wavelength and fluorescence intensity can be used as axes in the fluorescence emission spectra. The maximum emission wavelength and fluorescence intensity of the residues (such as tryptophan and tyrosine) show a close relation to the polarities of their microenvironment. Fig.9 shows the 3D fluorescence spectra for BSA, indigotin-BSA complex, HSA and indigotin-HSA complex, and the corresponding characteristic parameters of four systems are listed in Table 6.
Fig.8 Synchronous fluorescence spectra of (A) BSA and (B) HSA with different concentrations of indigotin at Δλ=15 nm (a) and Δλ=60 nm (b), [BSA]=[HSA]=5.0 μM, Cindigotin (1-6)=0.0, 4.0, 8.0, 12.0, 16.0 and 20.0 μM. T=298 K.

Fig.9 Three-dimensional fluorescence spectra of BSA (1), BSA-indigotin system (2), HSA (3), and HSA-indigotin system (4);conditions: c(BSA)=c(HSA)=5.0 μM, c(indigotin)=16.0 μM, pH=7.4, T=298 K.
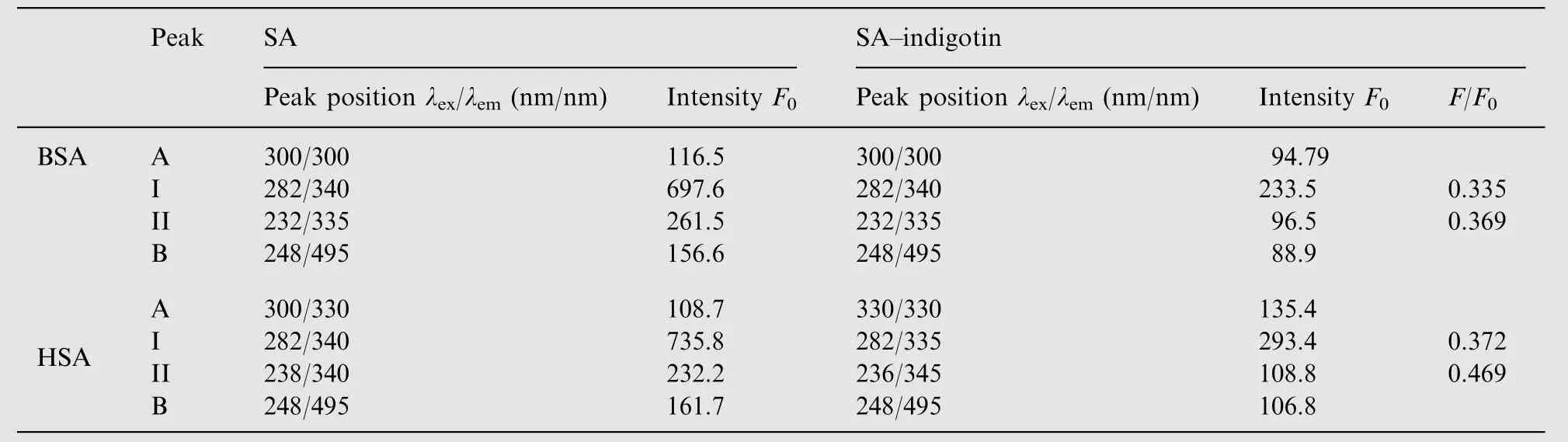
Table 6 Three-dimensional fluorescence characteristic parameters of SA and the indigotin-SA systems.
Four peaks can be seen in the 3D fluorescence spectra(Fig.9). The peak at the left (Peak A) is the first-ordered Rayleigh scattering peak, whose emission wavelength equals the excitation wavelength,whereas the peak at the right(Peak B) is the second-ordered Rayleigh scattering peak, whose emission wavelength is two times as the excitation. The fluorescence intensity of peak A increased with the addition of indigotin.It could be attributed to indigotin-HSA complex that made the volume of HSA increased, which in turn resulted in the enhancement of the scattering effect.
There are two strong fluorescence peaks seen in the middle of Fig.9, Peak I mainly reflects the spectral behavior of tryptophan and tyrosine residues on BSA/HSA;Peak II shows the behavior of polypeptide backbone structures of BSA/HSA and its intensity is correlated with the secondary structures of proteins [41]. The fluorescence intensities of Peak I and II for the indigotin-BSA complex have been quenching about 66.5%and 63.1%(Table 6);for the indigotin-HSA complex,its Peak I and II have been quenching about 62.8% and 53.1%,moreover,their corresponding maximum emission wavelength slightly blue shifted (from 340 to 335 nm) and red shifted(from 340 to 345 nm), respectively. This indicated that the interactions between indigotin and proteins induced a slight unfolding of the polypeptide backbone in BSA/HSA, and indigotin would locate at the vicinity of tryptophan residues on BSA/HSA. The results were in accordance with those obtained by the synchronous fluorescence spectra, site marker competitive experiments and CD spectra studies.
3.6.3. FT-IR spectra studies
The interactions between indigotin and BSA/HSA were characterized by infrared spectroscopy. Because there was no major spectral shifting for BSA/HSA amide I band at 1656/1655 cm-1(mainly C=O stretch) and amide II band at 1548/1547 cm-1(C-N stretching coupled with N-H bending modes) in the presence of indigotin, the difference spectra[(protein solution+indigotin solution)-(protein solution)]were obtained in order to monitor the intensity changes of these vibrations, and their results are shown in Figs. 10 and 11.
When the molar ratio of HSA/BSA to indigotin was maintained at 1:3, the intensity changes were observed for BSA/HSA amide I band at 1650/1654 cm-1and amide II band at 1547/1549 cm-1,and negative features were observed in the difference spectra for the amide I and II bands at 1659 and 1547 cm-1(BSA), 1655 and 1547 cm-1(HSA) in the BSAindigotin and HSA-indigotin systems, respectively (Figs. 10 and 11). For the BSA-indigotin and HSA-indigotin complexes, the intensities of the amide I band of BSA/HSA at 1656/1655 cm-1decreased, which suggested that the percentage content of α-helical structure of proteins would decrease at high indigotin concentration [42-44]. The results were in accordance with the CD spectra data analysis.
3.6.4. CD spectra studies
Circular dichroism (CD) is an important technique in the structural characterization of proteins, and especially for secondary structure determination.Thus,in the current study,the CD method has been applied to monitor the secondary structural change of proteins upon interaction with ligands.The CD spectra of BSA/HSA in the absence and presence of indigotin are shown in Fig.12. As shown in Fig.12, proteins exhibited two negative ellipticities at 208 and 220 nm in the UV region, which were characteristic of the typical α-helix structure of proteins.The CD results are expressed in terms of mean residue ellipticity (MRE) by the following equation [5]:

where Cpis the molar concentration of protein, n is the number of amino acid residues and l is the path length. The a-helix contents of free and combined BSA and HSA are calculated from MRE values at 208 nm using the equation[5]:
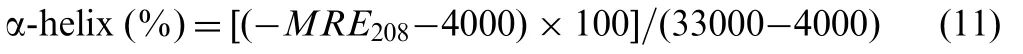
in Eq. (11), MRE208represents the observed MRE value at 208 nm,4000 is the MER value of the β-form and random-coil conformation cross at 208 nm,and 33000 is the MER value of a pure a-helix at 208 nm. According to the above equations,the a-helix content of BSA and HSA was calculated. It decreased from 51.7% in free BSA to 46.9% in indigotin-BSA, and from 57.0% in free HSA to 44.3% in indigotin-HSA.The decrease of α-helix contents indicated that indigotin bound to the amino acid residues of the main polypeptide chain of BSA/HSA and destroyed the hydrogen-networks [9].Furthermore,the CD spectra of BSA/HSA in the presence and absence of indigotin were observed to be similar in shape,indicating that the structure of proteins was also predominantly a-helical even after binding to indigotin [5]. So, we believed that the binding between indigotin and BSA/HSA induced the structural change of proteins.
3.7. Hydrophobic interactions
In order to examine the hydrophobic contact in the BSAindigotin and HSA-indigotin complexes,the antisymmetric and symmetric CH2stretching vibrations of proteins [44] were Investigated in the region 2800-3000 cm-1. The CH2bands of free BSA located at 2961, 2932 and 2861 cm-1shifted to 2956,2924 and 2854 cm-1(indigotin),and yet,the CH2bands of free HSA located at 2938 and 2874 cm-1shifted to 2917 and 2880 cm-1(indigotin) in the protein-dye complexes (Fig.13).So, the antisymmetric and symmetric CH2stretching vibrations of proteins shifted.The results indicated that the indigotin-BSA/HSA complexes were formed by the hydrophobic interaction force [42,44], which was consistent with fluorescence spectroscopic results discussed above.

Fig.10 FT-IR spectra in the region of 1800-900 cm-1 for free BSA (0.10 mM), the BSA-indigotin complex (the molar ratio of BSA/indigotin was maintained at 1:3), and its corresponding difference spectra (indicated in the figure). The contribution of indigotin was subtracted from different spectra in this region.
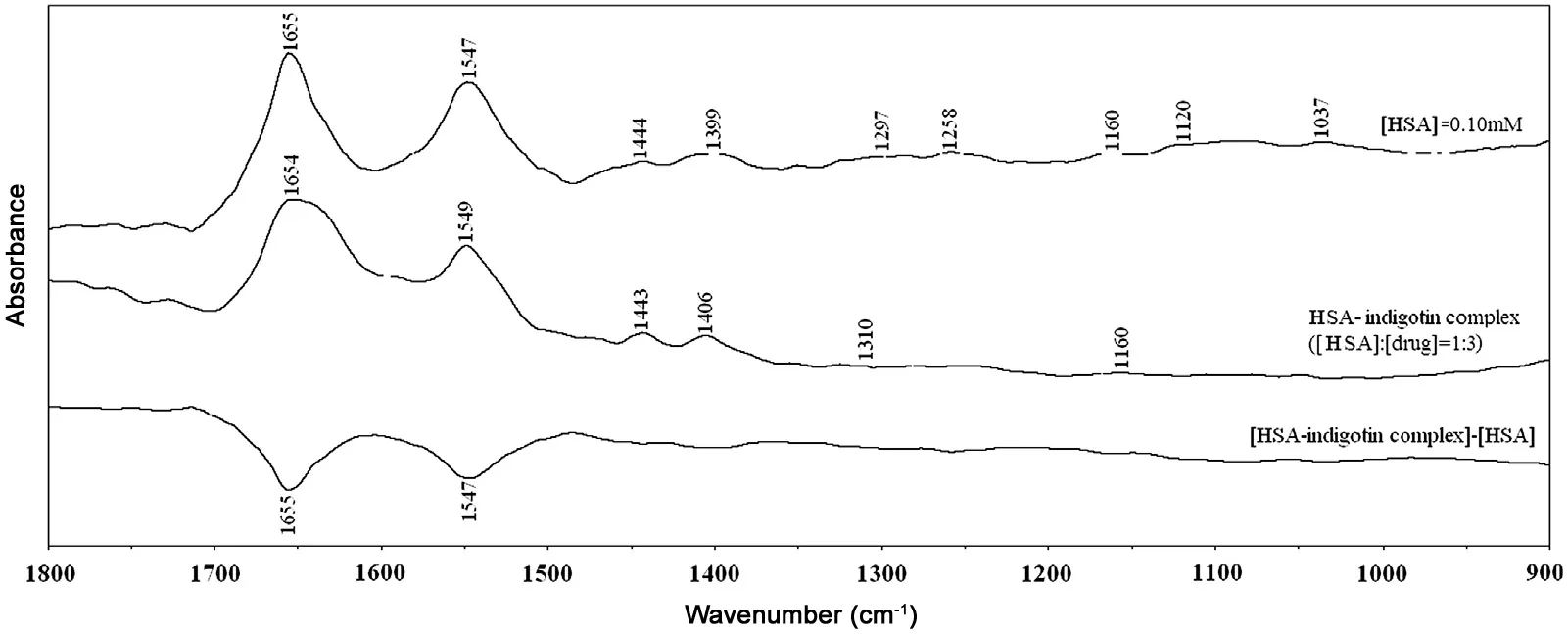
Fig.11 FT-IR spectra in the region of 1800-900 cm-1 for free HSA (0.10 mM), the HSA-indigotin complex (the molar ratio of HSA/indigotin was maintained at 1:3), and its corresponding difference spectra (indicated in the figure). The contribution of indigotin was subtracted from different spectra in this region.

Fig.12 CD spectra of BSA(A)and HSA(B)by indigotin.Conditions:c(BSA)=c(HSA)=1.0 μM;c(indigotin)a-c,0,10.00,20.00 μM.
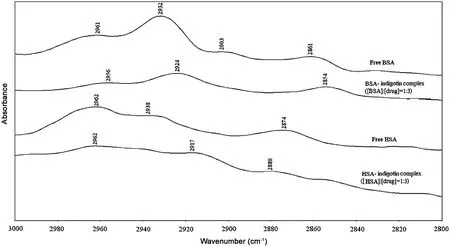
Fig.13 Infrared spectra of proteins CH2 symmetric and antisymmetric stretching vibrations and indigotin-BSA/HSA complexes in the region 3000-2800 cm-1, the contribution of the CH2 stretching vibrations of indigotin was subtracted in the region.
4. Conclusions
In this paper, we have investigated the binding interactions of proteins with indigotin at simulative physiological condition using various methods. The experimental results showed that indigotin could bind to SAs to form a indigotin-SA complex with one binding site. From the thermodynamic parameters calculation, it suggested that the indigotin-SAs complexes were formed by the hydrophobic interaction force.The results of synchronous fluorescence, 3D fluorescence and CD spectra indicated that the microenvironment of tyrosine and tryptophan residues on proteins altered with the addition of indigotin. The study would be useful for understanding the biological effects and functions of dyes in the body. In addition, the comparisons of binding affinity between SAs and indigotin derivatives or other azo dyes are worthy to be explored in the future because they are likely to provide useful information for studying their toxicity at the functional macromolecular level.
The author gratefully acknowledges financial support by the Education Department of Sichuan Province (12ZA171).
[1] E. Sales, R. Kanhonou, C. Baixauli, et al., Sowing date,transplanting, plant density and nitrogen fertilization affect indigo production from Isatis species in a Mediterranean region of Spain, Ind. Crop. Prod. 23 (2006) 29-39.
[3] S.M.T. Shaikh, J. Seetharamappa, S. Ashoka, et al., A study of the interaction between bromopyrogallol red and bovine serum albumin by spectroscopic methods, Dyes Pigm. 73 (2007)211-216.
[4] A. Sulkowska, Interaction of drugs with bovine and human serum albumin, J. Mol. Struct. 614 (2002) 227-232.
[5] A.Z. Wu, C.Z. Lin, Y.J. Zhai, Investigation of the interaction between two phenylethanoid glycosides and bovine serum albumin by spectroscopic methods, J. Pharm. Anal. 3 (2013) 61-65.
[6] P.B.Kandagal,S.Ashoka,J.Seetharamappa,et al.,Study of the interaction of an anticancer drug with human and bovine serum albumin: spectroscopic approach, J. Pharm. Biomed. Anal. 41(2006) 393-399.
[7] J. Chamani, H. Vahedian-Movahed, M.R. Saberi, Lomefloxacin promotes the interaction between human serum albumin and transferrin: a mechanistic insight into the emergence of antibiotic's side effects Original Research Article, J. Pharm. Biomed.Anal. 55 (2011) 114-124.
[8] P.N. Naik, S.A. Chimatadar, S.T. Nandibewoor, Interaction between a potent corticosteroid drug-dexamethasone with bovine serum albumin and human serum albumin: a fluorescence quenching and fourier transformation infrared spectroscopy study, J. Photochem. Photobiol. B 100 (2010) 147-159.
[9] F.L.Cui,J.Fan,J.P.Li,et al.,Interactions between 1-benzoyl-4-p-chlorophenyl thiosemicarbazide and serum albumin:investigation by fluorescence spectroscopy, Bioorg. Med. Chem.12 (2004) 151-157.
[10] S. Nafisi, A. Panahyab, G.B. Sadeghi, Interactions between β-carboline alkaloids and bovine serum albumin: investigation by spectroscopic approach, J. Lumin. 132 (2012) 2361-2366.
[11] X.R.Pan,P.F.Qin,R.T.Liu,et al.,Characterizing the interaction between tartrazine and two serum albumins by a hybrid spectroscopic approach, J. Agric. Food Chem 59 (2011) 6650-6656.
[12] D.C. Carter, J.X. Ho, Structure of serum albumin, Adv. Protein Chem. 45 (1994) 153-203.
[13] D. Shcharbin, M. Janicka, M. Wasiak, et al., Serum albumins have five sites for binding of cationic dendrimers, Biochim.Biophys. Acta 1774 (2007) 946-951.
[14] P. Banerjee, S. Ghosh, A. Sarkar, et al., Fluorescence resonance energy transfer: a promising tool for investigation of the interaction between 1-anthracene sulphonate and serum albumins,J. Lumin. 131 (2011) 316-321.
[15] Y.J. Hu, Y. Liu, X.H. Xiao, Investigation of the interaction between berberine and human serum albumin, Biomacromolecules 10 (2009) 517-521.
[16] D. Yuan, Z.L. Shen, R.T. Liu, et al., Study on the interaction of La3+with bovine serum albumin at molecular level, J. Lumin.131 (2011) 2478-2482.
[17] N. Shahabadi, M. Mohammadpour, Study on the interaction of sodium morin-5-sulfonate with bovine serum albumin by spectroscopic techniques, Spectrochim. Acta Part A 86 (2012) 191-195.
[18] M.R. Eftink, C.A. Ghiron, Fluorescence quenching studies with proteins, Anal. Biochem. 114 (1981) 199-227.
[19] X.L. Wei, J.B. Xiao, Y.F. Wang, et al., Which model based on fluorescence quenching is suitable to study the interaction between trans-resveratrol and BSA?, Spectrochim. Acta Part A 75 (2010) 299-304.
[20] M.X. Xie, X.Y. Xu, Y.D. Wang, Interaction between hesperetin and human serum albumin revealed by spectroscopic methods,Biochim. Biophys. Acta 2005 (1724) 215-224.
[21] Y.X. Wang, L. Li, L.J. Sheng, et al., Spectroscopic study on the inherent binding information of cationic perfluorinated surfactant with bovine serum albumin, J. Fluorine Chem. 132 (2011) 489-494.
[22] B. Ojha, G. Das, The Interaction of 5-(Alkoxy)naphthalen-1-amine with bovine serum albumin and Its effect on the conformation of protein, J. Phys. Chem. B 114 (2010) 3979-3986.
[23] R. Sivakumar, S. Naveenraj, S. Anandan, Interactions of serum albumins with antitumor agent benzo [a] phenazine—a spectroscopic study, J. Lumin. 131 (2011) 2195-2201.
[24] H. Wu, X.J. Zhao, P. Wang, et al., Electrochemical site marker competitive method for probing the binding site and binding mode between bovine serum albumin and alizarin red S, Electrochim. Acta 56 (2011) 4181-4187.
[25] J.S. Mandeville, H.A. Tajmir-Riahi, Complexes of dendrimers with bovine serum albumin, Biomacromolecules 11 (2010) 465-472.
[26] Y.P. Zhang, S.Y. Shi, X.Q. Chen, et al., Investigation on the Interaction between Ilaprazole and bovine serum albumin without or with different C-ring flavonoids from the viewpoint of food_drug interference, J. Agric. Food Chem. 59 (2011)8499-8506.
[27] M. Bhattacharya, N. Jain, S. Mukhopadhyay, Insights into the mechanism of aggregation and fibril formation from bovine serum albumin, J. Phys. Chem. B 115 (2011) 4195-4205.
[28] X.Y. Yu, R.H. Liu, F.X. Yang, et al., Study on the interaction between dihydromyricetin and bovine serum albumin by spectroscopic techniques, J. Mol. Struct. 985 (2011) 407-412.
[29] D. Charbonneau, M. Beauregard, H.A. Tajmir-Riahi, Structural analysis of human serum albumin complexes with cationic lipids,J. Phys. Chem. B 113 (2009) 1777-1784.
[30] Y.Y. Yue, X.G. Chen, J. Qin, et al., Characterization of interaction between C.I. acid green 1 and human serum albumin: spectroscopic and molecular modeling method, Dyes Pigm. 83 (2009) 148-154.
[31] D.P.Ross,S. Subramanian,Thermodynamics of protein association reactions: forces contributing to stability, Biochemistry 20(1981) 3096-3102.
[32] J. Zhang, X.J. Wang, Y.J. Yan, et al., Comparative Studies on the Interaction of Genistein, 8-Chlorogenistein, and 3', 8-Dichlorogenistein with Bovine Serum Albumin, J. Agric. Food Chem. 59 (2011) 7506-7513.
[34] S.M.T.Skaikh, J. Seetharamappa,P.B. Kandagal,et al., Binding of the bioactive component isothipendyl hydrochloride with bovine serum albumin, J. Mol. Struct. 786 (2006) 46-52.
[35] J.R. Lakowicz, G. Weber, Quenching of fluorescence by oxygen:A probe for structural fluctuation in macromolecules, Biochemistry 12 (1973) 4161-4170.
[36] A. Samanta, B.K. Paul, N. Guchhait, Spectroscopic probe analysis for exploring probe-protein interaction: a mapping of native, unfolding and refolding of protein bovine serum albumin by extrinsic fluorescence probe, Biophys. Chem. 156 (2011)128-139.
[37] X.J. Guo, A.J. Hao, X.W. Han, et al., The investigation of the interaction between ribavirin and bovine serum albumin by spectroscopic methods, Mol. Biol. Rep. 38 (2011) 4185-4192.
[38] Y.Y. Yue, J.M. Liu, J. Fan, et al., Binding studies of phloridzin with human serum albumin and its effect on the conformation of protein, J. Pharm. Biomed. Anal. 56 (2011) 336-342.
[39] Z.J. Cheng, Y.T. Zhang, Spectroscopic investigation on the interaction of salidroside with bovine serum albumin, J. Mol.Struct. 889 (2008) 20-27.
[40] D.J. Li, Y. Wang, J.J. Chen, et al., Characterization of the interaction between farrerol and bovine serum albumin by fluorescence and circular dichroism, Spectrochim. Acta Part A 79 (2011) 680-686.
[41] F.F. Tian, F.L. Jiang, X.L. Han, et al., Synthesis of a novel hydrazone derivative and biophysical studies of its interactions with bovine serum albumin by spectroscopic, electrochemical, and molecular docking methods, J. Phys. Chem. B 114 (2010) 14842-14853.
[42] P. Bourassa, C.D. Kanakis, P. Tarantilis, et al., Resveratrol,genistein, and curcumin bind bovine serum albumin, J. Phys.Chem. B 114 (2010) 3348-3354.
[43] Z.J. Cheng, Comparative studies on the interactions of honokiol and magnolol with human serum albumin, J. Pharm. Biomed.Anal. 66 (2012) 240-251.
[44] S. Dubeau, P. Bourassa, T.J. Thomas, et al., Biogenic and synthetic polyamines bind bovine serum albumin, Biomacromolecules 11 (2010) 1507-1515.
 Journal of Pharmaceutical Analysis2013年4期
Journal of Pharmaceutical Analysis2013年4期
- Journal of Pharmaceutical Analysis的其它文章
- Quantitative analysis of cefixime via complexation with palladium(II) in pharmaceutical formulations by spectrophotometry
- Simultaneous pharmacokinetic assessment of cefadroxil and clavulanic acid in human plasma by LC-MS and its application to bioequivalence studies
- Application of UPLC-MS/MS for separation and quantification of 3α-Hydroxy Tibolone and comparative bioavailability of two Tibolone formulations in healthy volunteers
- A simple method for the isolation and purification of resveratrol from Polygonum cuspidatum
- Fingerprint analysis of Cirsium japonicum DC. using high performance liquid chromatography
- Ultra-high-performance liquid chromatography for the determination of exenatide in monkey plasma by tandem quadrupole mass spectrometry