
The Mechanism of the Acclimation of Nannochloropsis oceanica to Freshwater Deduced from Its Transcriptome Profiles
2015-03-15 01:44:02GUOLiandYANGGuanpin
GUO Li, and YANG Guanpin,
1) Laboratory of Marine Genetics and Breeding, College of Marine Life Sciences, Ocean University of China, Qingdao 266003, P. R. China
2) Institute of Evolution and Marine Biodiversity, Ocean University of China, Qingdao 266003, P. R. China
The Mechanism of the Acclimation of Nannochloropsis oceanica to Freshwater Deduced from Its Transcriptome Profiles
GUO Li1), and YANG Guanpin1),2),*
1) Laboratory of Marine Genetics and Breeding, College of Marine Life Sciences, Ocean University of China, Qingdao 266003, P. R. China
2) Institute of Evolution and Marine Biodiversity, Ocean University of China, Qingdao 266003, P. R. China
? Ocean University of China, Science Press and Spring-Verlag Berlin Heidelberg 2015
In this study, we compared the transcriptomes of Nannochloropsis oceanica cultured in f/2 medium prepared with seawater and freshwater, respectively, aiming to understand the acclimation mechanism of this alga to freshwater. Differentially expressed genes were mainly assigned to the degradation of cell components, ion transportation, and ribosomal biogenesis. These findings indicate that the algal cells degrade its components (mainly amino acids and fatty acids) to yield excessive energy (ATP) to maintain cellular ion (mainly K+and Ca2+) homeostasis, while the depletion of amino acids and ATP, and the reduction of ribosomes attenuate the protein translation and finally slow down the cell growth.
Nannochloropsis oceanica; acclimation; ion depletion; ribosome biogenesis; protein translation
1 Introduction
Most species of genus Nannochloropsis inhabit seawater (Fawley et al., 2007), and are rich in proteins and eicosapentaenoic acid (EPA). When nitrogen is limited, they may accumulate fatty acids to as high as 60% of dry biomass (Rodolfi et al., 2009). Nannochloropsis are monoploid (Galloway, 1990; Kilian et al., 2011; Pan et al., 2011) and reproduce asexually (Pan et al., 2011). They are cultured as human food and animal feed additives (Hoffmann et al., 2010), and may be employed as biodiesel producers because they multiply fast and are rich in lipids (Rodolfi et al., 2009; Sukenik et al., 2009). With the development of diverse transformation tools, e.g., random integration (Chen et al., 2008), Agrobacteriummediated transformation (Cha et al., 2011), and homologous recombination (Kilian et al., 2011), as well as the accumulation of data, e.g., expressed sequence tags (EST) (Liang et al., 2013; Shi et al., 2008) and genome sequences (Carpinelli et al., 2014; Pan et al., 2011; Radakovits et al., 2012; Vieler et al., 2012; Wang et al., 2014), they have been evolving as the models of industrial microalgae. Recently, Nannochloropsis have been considered as green yeasts as they are easy to culture and feasible for genetic manipulation (Weeks, 2011).
Nannochloropsis can survive and maintain slow growth in freshwater (Huang and Hu, 2013). As we have found inthe genome of N. oceanica (Pan et al., 2011), Nannochloropsis are extremely high in the content of microsatellites, thus may survive in freshwater environment by modifying the length of microsatellite motifs. Different mechanisms may underline the length variation of microsatellites, while slipped strand mispairing (SSM) mechanism is widely accepted. According to SSM, the insertion/deletion (Levinson et al., 1987), and gene expression regulation can be caused by terminating translation and peptide elongation, fusing ORF and separating ORF, hindering initiation of transcription, and changing the structure of DNA and RNA. However, among about 1000 microsatellites we have tested, no SSM was observed between Nannochloropsis cultured in seawater and freshwater, and more than 1000 microsatellites were not analyzable due to the difficulty of primer designing (data not shown). We proposed that SSM of microsatellites may not be the mechanism underlining the survival of N. oceanica in freshwater. A new way should be paved in order to understand the acclimation of N. oceanica to freshwater.
An organism may respond to environmental change through acclimation, or adaptation, or both. During the acclimation, cells regulate the expression of their genes slightly and change their traits plastically. In contrast, during the adaptation, cells will deeply regulate the expression of their genes and fix their performance. However, adaptation is time-consuming, which involves permanent change of DNA sequences. In a protist population, these two mechanisms may function concertedly (Sunday et al., 2014). It is common to believe that adaptationneeds hundreds of generations of cell division so that pre-existing mutants can evolve into the dominants in a population; while acclimation plays a key role during a short term response. Epigenetic shifting is one of the regulating ways of gene expression (Crevillen et al., 2014). During the acclimation, such shifting should happen less frequently. Therefore, Nannochloropsis may acclimate to freshwater fast by changing the expression of their genes temporarily if less generations of cellular division are allowed (Lohbeck et al., 2012).
RNA sequencing (RNA-seq), also called whole transcriptome shotgun sequencing, is a technology that uses the capabilities of a massively parallel sequencing tools to profile the transcripts abundance either temporarily or spatially or both (Wang et al., 2009). Application of this method has already altered our view of the extent and the complexity of eukaryotic transcriptomes, for example, the precise abundance of transcripts and their isoforms, the sequence of previously annotated 5’ and 3’ ends of genes and the boundary of exons and introns (Mortazavi et al., 2008; Trapnell et al., 2010; Trapnell et al., 2012). In addition, RNA-seq may also reveal the alternative splicing, a widespread process of regulating gene expression temporarily and spatially (Blencowe, 2006; Fedor, 2008; Stamm et al., 2005).
In this study, we compared the transcriptomes of N. oceanica cultured in f/2 medium prepared with seawater and freshwater, respectively. We aimed to understand the physiologic mechanism underlining its acclimation to freshwater, and reveal the biological processes correlated with the salinity acclimation.
2 Materials and Methods
2.1 N. oceanica and Its Culturing
N. oceanica was provided by Key Laboratory of Mariculture of Chinese Ministry of Education, Ocean University of China, which was cultured in f/2 (pH 7.8, salinity 30) (Guillard and Ryther, 1962; Guillard, 1975) and SE media. The f/2 medium was prepared with seawater near Qingdao shore which was filtrated through a membrane with pores of 0.4 μm in diameter ahead of use. SE medium was modified from Bristol medium by supplying soil extract as was described online (http://web. biosci.utexas.edu/utex/default.aspx). Both media were autoclaved at 121℃ for 30 min. The alga was cultured at 25℃ and under an irradiation of 70 μmol s-1m-2following a rhythm of 12 h dark:12 h light.
2.2 RNA Extraction, Library Construction and Sequencing
The cells were harvested at the logarithm growth phase (about (2–5)×107cell mL-1) through centrifugation at 4000g for 10 min. Then the cells were washed once with sterilized seawater, suspended in seawater and divided into aliquots of 1.5 mL in volume, precipitated into pellets by centrifuging at 10000 g for 2 min, frozen immediately in liquid nitrogen and stored at -80℃ for RNA extraction. The total RNA was extracted using Trizol Reagent (Invitrogen, USA) following manufacturer’s instructions. The extracted RNA was assessed for any degradation and contamination through electrophoresis in 1% agarose gel. The concentration of RNA was measured on a Qubit2.0 Fluorometer. The purity and integrity of RNA was determined with a NanoDrop Spectrophotometer and an Agilent 2100 Bioanalyzer, respectively.
About 3 μg RNA with RNA integrity (RIN) value of about 8 was used for library construction. Poly (A) tailed mRNA was isolated using the oligo(dT) magnetic beads in Oligotex mRNA Kits (Qiagen), fragmented and used as the template for first-strand cDNA synthesis with reverse transcriptase and random hexamers. The second-strand cDNA was synthesized with RNase H and DNA polymerase I. Following end polishing, each of the ds cDNA fragments was added a single ‘A’ base and ligated with adapters. The modified ds cDNA fragments were gel purified and PCR amplified, yielding a cDNA sequencing library which was quantified with an Agilent 2100 Bioanalyzer.
Transcriptome sequencing was carried out on an Illumina HiSeqTM2000 platform which generates about 100 bp paired-end (PE) raw reads (Novogene Bioinformatics Technology Co. Ltd). These raw reads in FASTA format were processed through in-house perl scripts, yielding a clean reads set. During the processing, GC content and quality parameters including Q20 and Q30 were calculated. All the downstream analyses were based on the clean reads sets.
The gene models predicted from the genome were downloaded from NCBI (Vieler et al., 2012) and used as reference transcripts. An index of the reference transcripts was built using Bowtie v0.12.8 (Alverson, 2007). The clean reads were aligned onto the reference transcripts using TopHat v2.0.7 (Trapnell et al., 2012). We preferred TopHat to other tools during such an alignment as it generates a file of splice junctions. The Cufflinks v2.1.1 Reference Annotation Based Transcript (RABT) assembly method was used to construct and identify both known and novel transcripts from the reads remained of TopHat alignment.
2.3 Determination of Differential Transcripts
Prior to determination of differential transcripts, the read counts were adjusted for each sequenced library with edgeR program package through one scaling normalized factor. HTSeq v0.5.3 (http://www-huber.embl.de/users/ anders/HTSeq) was used to count the reads mapped to each gene. The abundance of each transcripts was normalized into RPKM (reads per kilobase of exon model per million mapped reads), the most commonly used parameter indicating the abundance of a transcript (Mortazavi et al., 2008). Differential transcripts between freshwater and seawater were determined with DESeq R package (1.12.0) (Anders et al., 2010). The resulted p-values were adjusted with Benjamini and Hochberg’s method (Benjamini et al., 1995). The adjusted p-value of 0.005 and log2(Foldchange) of 1.00 were set as thethresholds for differential transcripts.
2.4 GO and KEGG Enrichment of Differential Transcripts
The differential transcripts were enriched into Gene Ontology (GO) terms (function groups) with GOseq R Package (Young et al., 2010), and the gene length bias was corrected. GO terms with corrected p-value of < 0.05 were considered significant. These differential transcripts were also enriched into KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways (http://www.genome.jp/ kegg/) with KOBAS software (Mao et al., 2005).
3 Results
3.1 Algal Culture
When N. oceanica was cultured in f/2 medium prepared with seawater, it maintained a logarithm growth around day 8. The algal cells were harvested and inoculated into f/2 medium prepared with seawater and freshwater, respectively. It was found that the alga recovered a logarithm growth in about 8 days in f/2 mediumprepared with seawater, but lived and grew slowly in f/2 medium prepared with freshwater. When the algal cells in f/2 medium prepared with freshwater were transferred back to f/2 medium prepared with seawater, they recovered a logarithm growth fast (Fig.1).
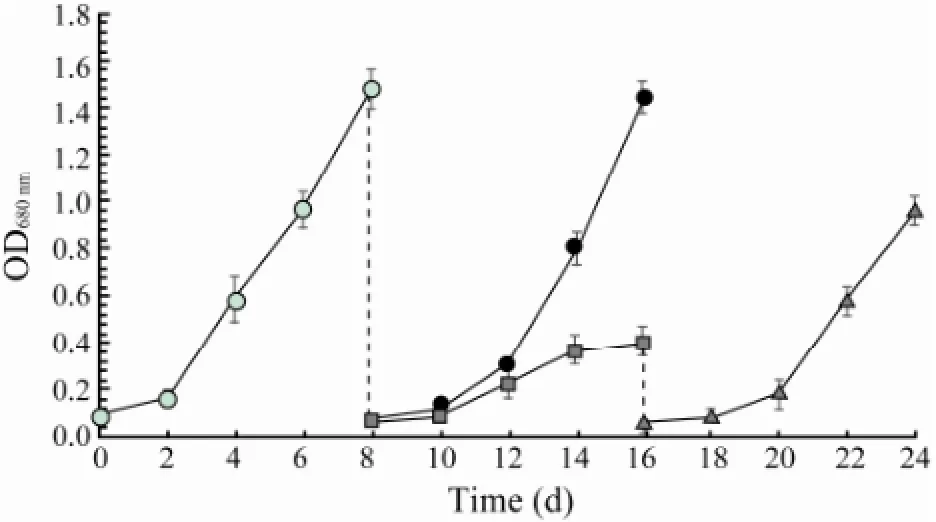
Fig.1 The growth of N. oceanica in f/2 medium prepared with seawater and freshwater, respectively. N. oceanica was cultured in f/2 medium prepared with seawaterwhich was harvested at the end of logarithm growth phase and cultured again either in f/2 medium prepared with seawateror in f/2 medium prepared with freshwaterThe algal cells cultured in f/2 medium prepared with freshwater were harvested again and cultured in f/2 medium prepared with seawater
3.2 Transcriptome Sequencing
In total, 81274952 and 78221736 raw reads were generated for N. oceanica cultured in freshwater and seawater, respectively, on an Illumina HiSeqTM2000 platform. After trimming adaptors and removing low quality reads, 77824056 (Q20> 96.97%; 7.78 GB in total length) and 74796988 (Q20> 96.92%; 7.48 GB in total length) quality (clean) reads were obtained for N. oceanica cultured in freshwater and seawater, respectively. Of the clean reads, 82.03% and 82.84% were stacked on the gene models, respectively, and 3.70% and 3.66% were crossly mapped to different gene models, respectively. Except for the gene models cited and used as references, 2025 novel transcripts which were not predicted from the genome reference were also assembled and used as the transcripts. In total, 14838 transcripts were obtained from clean reads. Of them, 13644 (91.95%) and 14112 (95.11%) were from N. oceanica cultured in freshwater and seawater, respectively. These transcripts, both the cited and the newly assembled, were compared for their abundance in N. oceanica cultured in freshwater and seawater, respectively.
3.3 Differentially Expressed Genes (DEGs) Between Freshwater and Seawater Culture
Of 14838 genes, 704 were found to be differentially expressed in N. oceanica cultured in freshwater and seawater, respectively (log2(Foldchange) > 1, q-value < 0.005). Of the DEGs, 477 were up-regulated and 227 were downregulated when N. oceanica was shifted from seawater to freshwater. The DEGs with unknown function will reduce the transparency of the transcriptome; however, those with known function will certainly aid to understanding the physiological mechanism underlining the acclimation of N. oceanica to freshwater. Of 14838 genes, 7944 were annotated, i.e., these genes encode proteins with knownfunction. It was found that some of DEGs with known function were significantly enriched in 9 GO terms which annotated, i.e., these genes encode proteins with known belong to 3 major functional groups, including cellular component, molecular function and biological process. It was interesting to note that 3 GO terms (0003735, 0005840 and 0030529) contained a set of ribosomal proteins, and 2 GO terms (0043228 and 0043232) contained a set of non-membrane bounded organelle proteins. Others GO terms contained proteins functioning either intranslation and structural molecular activity or as cytoplasmic parts (Table 1).

Table 1 Differentially expressed genes (DEGs) significantly enriched in 9 GO terms
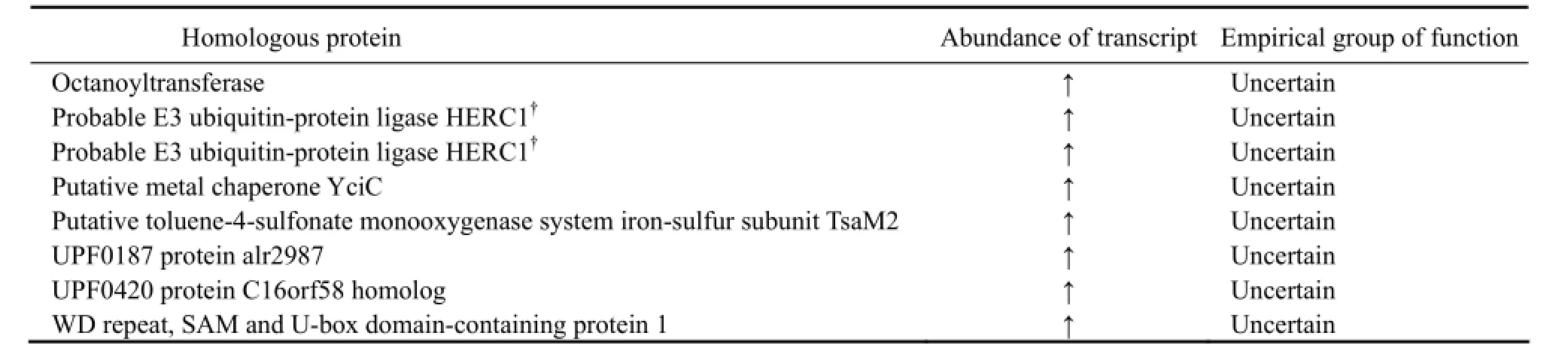
(continued)
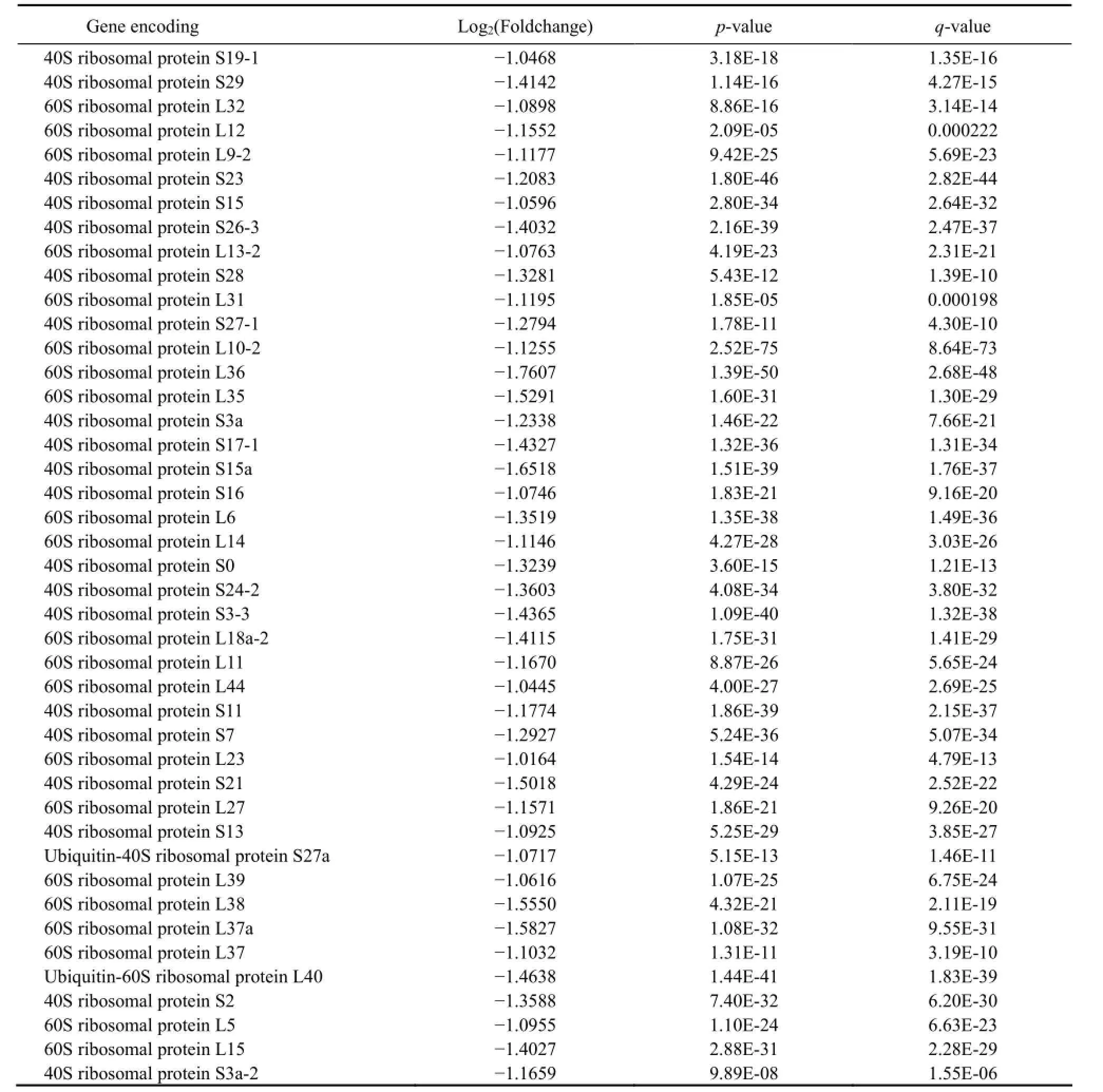
Table 3 Forty three ribosomal proteins encoded by DEGs down-regulated in freshwater and significantly enriched in GO terms
In order to understand the function of DEGs better, the function-known proteins enriched in 9 GO terms were empirically grouped (by referring to either literatures or information available online) into ion transportation; central metabolism pathways; protein translation; cell skeleton and division; protein, DNA and RNA modification; cell damage repairing; cell wall construction among others (Table 2); and ribosomal proteins (Table 3).
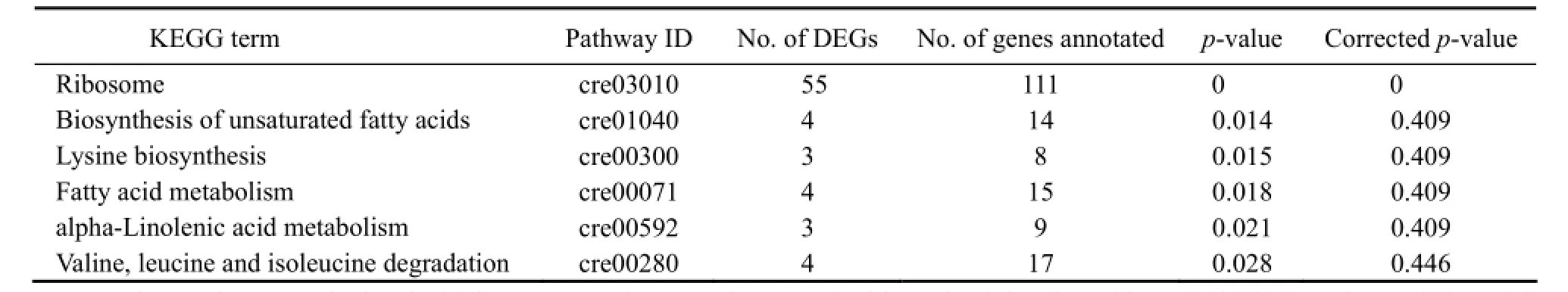
Table 4 DEGs enriched significantly in 6 KEGG pathways
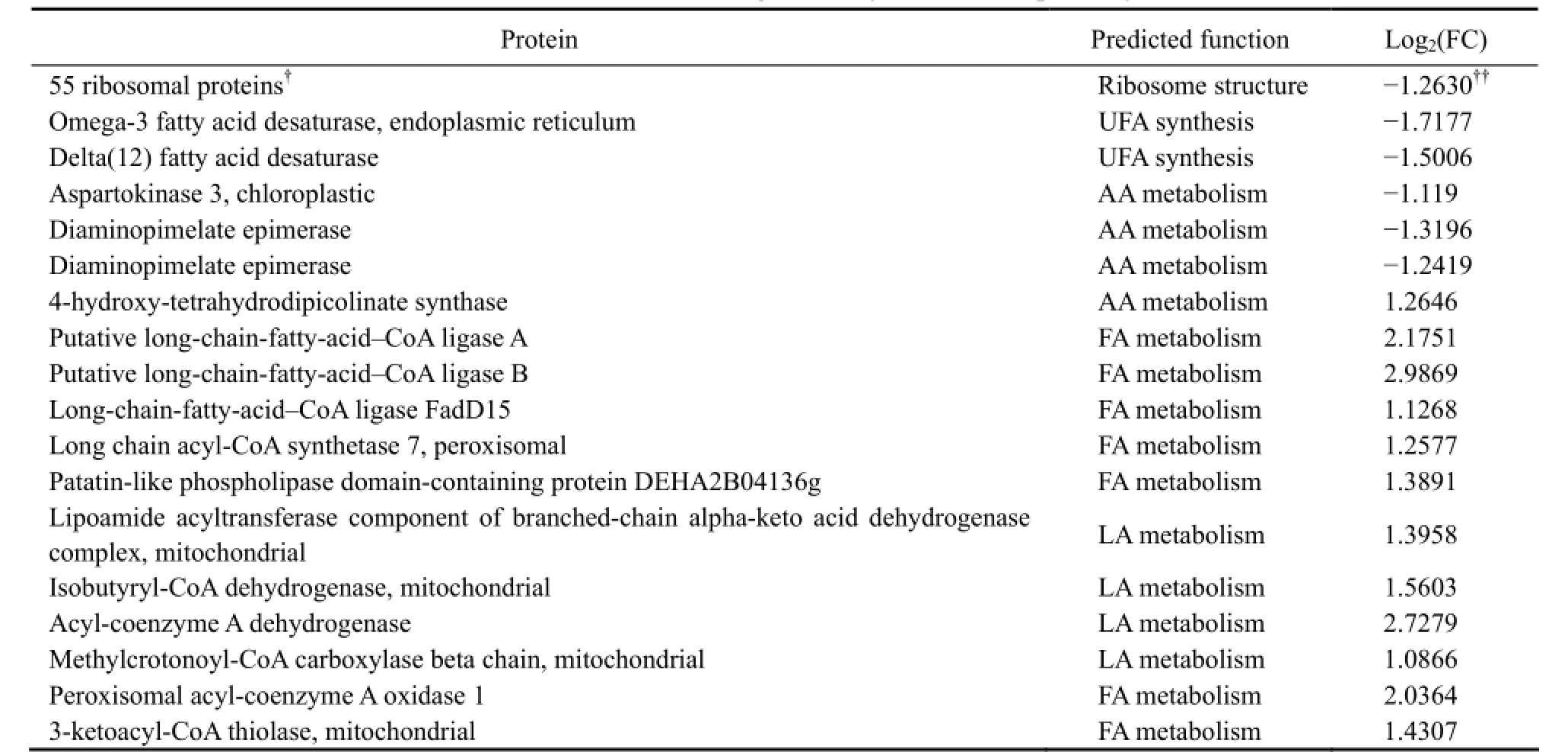
Table 5 Proteins enriched significantly in 6 KEGG pathways
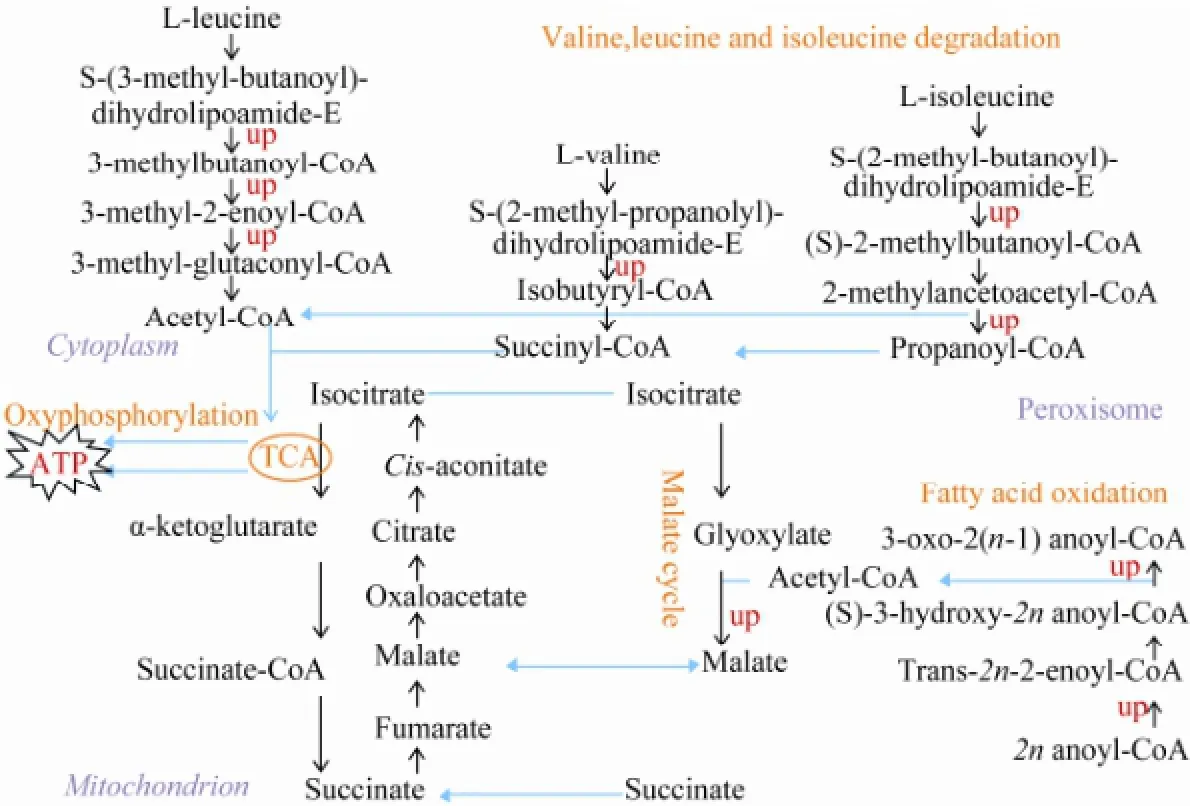
Fig.2 The up-regulated expression and compartmentalization of key enzyme-encoding genes involved in fatty acid and amino acid degradations.
GO term enrichment of DEGs has aided to an empirical understanding of the cellular response of N. oceanica to freshwater. In order to study the response, the DEGs were also enriched into KEGG metabolic pathways. In total, 73 DEGs were enriched in 6 pathways which included ribosome biogenesis; degradation of unsaturated fatty acids, lysine and fatty acid; alpha-linolenic acid metabolism; and valine, leucine and isoleucine degradation (Tables 4 and 5). It was noticed that the enrichment may not be significant according to the corrected p-value, except for ribosome proteins, as the corrected p-value of the later 5 pathways was > 0.05. We accepted these enrichments as only about half of the gene models have been annotated. Very interestingly, of 111 annotated ribosome associating DEGs, 55 were enriched in a unique pathway, ribosome, significant in corrected p values.
Except for ribosome structural proteins, the enriched proteins also included the enzymes involved in the degradation of fatty acids (unsaturated fatty acids, fatty acids and linolenic acid) and amino acids (lysine, valine, leucine and isoleucine). These enzymes and the pathways they run lead to the degradation of amino acids and fatty acids, yielding acetyl-CoA and small intermediate molecules which may enter TCA and oxidative phosphorylation, generate the cellular power currency ATP (Fig.2).
4 Discussion
4.1 Ion Homeostasis and Energy Demand
During the early evolution of life in seawater, K+emerged as the major cation of plasma, maintaining the electroneutrality and osmotic equilibrium of cells, and was involved in diverse biochemical processes and protein functions (Rodriguez-Navarro and Rubio, 2006; Walker et al., 1996). Cytoplasm has an absolute requirement for K+, while Na+is toxic; however, vacuole may attenuate such toxicity by replacing K+with Na+partially, as Na+is not toxic and can undertake osmotic functions. This replacement reduces the total K+requirement, improving growth when K+is limited. Externally supplied Ca2+reduces the toxic effect of NaCl, presumably by facilitating K+/Na+selectivity. Ca2+may also signal cellular responses to environmental changes (Hasegawa et al., 2000). Transmembrane transport proteins that mediate ion fluxes across the plasma membrane and tonoplast include H+translocating ATPases and pyrophosphatases, Ca2+-ATPases and secondary active transporters and channels, which are driven by the H+-electrochemical potential gradients established by H+pumps.
A comparison between modified freshwater f/2 and standard seawater f/2 indicated that the former is lower in the content of sodium and chloride ions than the latter, and is deficient in potassium and calcium ions. When N. oceanica was cultured in the modified freshwater f/2, this evolutionary selection confined or influenced its growth performance. In order to satisfy the growth, N. oceanica must strengthen its ability of K+acquirement. As was demonstrated by transcriptome profiling, the expression of a few genes associating with ion transportation were up regulated.
As either transporters or channels, their actions should be driven by a huge amount of energy. Modified freshwater f/2 is deficient for K+, thus N. oceanica must enrich a trace amount of K+possibly available for its growth; otherwise it must stop growing when K+is completely unavailable. This can be accomplished by shifting its central metabolism pathways to energy making if sodium and chloride ions did not stress the growth of N. oceanica in modified freshwater f/2. The cost of maintaining the homeostasis of K+and Ca2+was much higher than the saved in pumping out sodium and chloride ions in standard seawater medium.
In this study, we found that the metabolism of unsaturated and normal fatty acids, linolenic acid, and the degradation of lysine, valine, leucine and isoleucine were increased. The malate cycle may also have been speeded up as the gene encoding malate synthase was up regulated when N. oceanica was cultured in modified freshwater f/2. These changes may orient cells to yield acetyl-CoA and small intermediate molecules which may enter into TCA and oxidative phosphorylation, generating NADH and ATP for satisfying the energy demand of cells, making cells amino acids depleted and ATP excessive.
4.2 Putative Mechanisms Underlining the Acclimation of N. oceanica to Freshwater
In modified freshwater f/2, N. oceanica lives but grows slowly, i.e., it acclimates to freshwater by gearing down its growth and proliferation so that the majority of cellular fuels (ATP and NADH) are burned to maintain ion homeostasis (mainly K+and Ca2+).
We found that the most down regulated and significantly enriched in both GO terms and KEGG pathways were a ribosomal protein set. Thus the ‘living but growing slowly’ strategy of N. oceanica in acclimating to freshwater was mediated by a mechanism functioning on these proteins. This is reasonable as ribosome biogenesis has clear links to cell growth (Lempiainen and Shore, 2009). Cell growth, or increase in cell mass, requires prodigious numbers of ribosomes, the molecular factories that carry out protein synthesis. This process is staggering in scale as it involves the coordinate regulation of all three nuclear RNA polymerases to produce roughly equimolar amounts of four ribosomal RNAs (rRNAs) and about 80 ribosomal proteins (RPs). In rapidly growing yeast cells, rRNA constitutes approximately 80% of total cellular nucleic acid, and nearly 50% of all RNA polymerase II (RNAP II) transcription initiation events occur on RP genes. In addition to the RNA and protein components of the ribosome itself, over 200 additional proteins and noncoding RNAs participate in the complex series of processing, modification, assembly, and nuclear importexport reactions required for producing cytoplasmic 60S and 40S particles (Loewith and Hall, 2011; Lempiainen and Shore, 2009). The production of all these abundant molecules represents a huge energetic investment. Notsurprisingly, the algal cells have to slow down their ribosome biogenesis as a large portion of energy has to be applied in maintaining ion homeostasis that associates with their survival. The reduction of ribosomal abundance will certainly slow down cellular anabolism, and finally limit the growth of cells and their proliferation.
To these understandings, we presumed that the algal cells degraded their components (mainly amino acids and fatty acids) to yield energy in order to maintain cellular ion (mainly K+and Ca2+) homeostasis. The depletion of amino acids and reduction of ribosomes decreased the protein translation, cell growth and division, which finally help N. oceanica acclimate to freshwater (Fig.3).
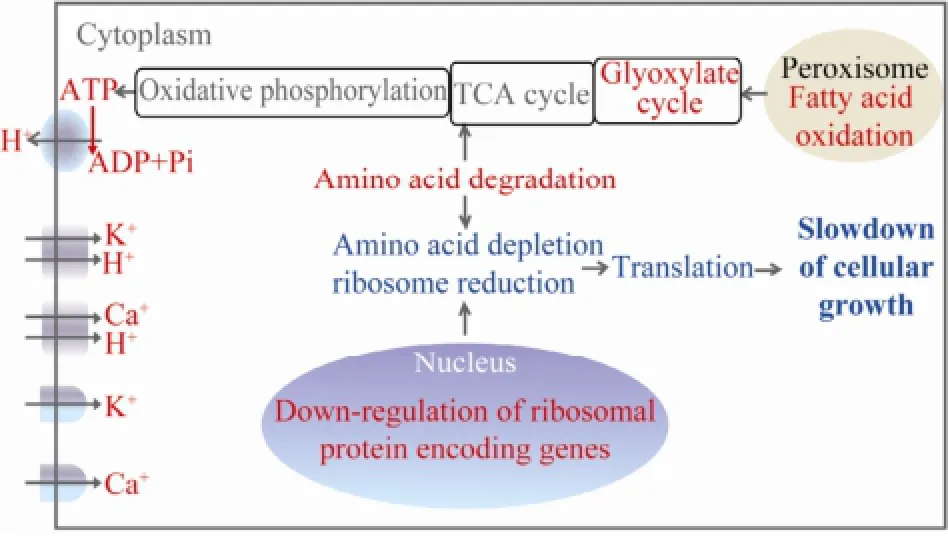
Fig.3 A presumed mechanism underlining the acclimation of N. oceanica to freshwater. The up-regulated cellular processes and components were in red while the downregulated cellular processes were in green.
4.3 The Significance and Future Research
N. oceanica is a unicellular microalga which can survive and grow slowly in freshwater. This may partially mimic the evolution of plants from seawater to freshwater, and then to territorial environment. Therefore, detailed documentation of the effectors mediating ion depletion and ribosome reduction may help to understand this important evolutionary process. N. oceanica may be a good biofuel producer, which has received an intensive attention from both researching community and industrial sector worldwide. Nannochloropsis are monoploid (Kilian et al., 2011; Pan et al., 2011; Galloway, 1990) and reproduce asexually (Pan et al., 2011). Diverse transformation tools (Cha et al., 2011; Chen et al., 2008; Huang et al., 2011; Kilian et al., 2011) have been developed for this alga, and the chemical and physical mutation methods have been proved to be very effective (Galloway, 1990; Ma et al., 2013). It is possible to genetically modify this alga to improve the growth performance and the content of desirable materials. With the acclimation and adaptation to freshwater, this alga may serve as a selector in artificial evolution for a new characteristic.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (31270408) and National High Technology Research and Development Program (863 Program) of China (2014AA022001).
Alverson, A. J., 2007. Strong purifying selection in the silicon transporters of marine and freshwater diatoms. Limnology and Oceanography, 52 (4): 1420-1429.
Anders, S., and Huber, W., 2010. Differential expression analysis for sequence count data. Genome Biology, 11: R106.
Benjamini, Y., and Hochberg, Y., 1995. Controlling the false discovery rate: A practical and powerful approach to multiple testing. Journal of the Royal Statistical Society (Series B), 57 (1): 289-300.
Blencowe, B. J., 2006. Alternative splicing: New insights from global analyses. Cell, 126: 37-47.
Carpinelli, E. C., Telatin, A., Vitulo, N., Forcato, C., D’Angelo, M., Schiavon, R., Vezzi, A., Giacometti, G. M., Morosinotto, T., and Valle, G., 2014. Chromosome scale genome assembly and transcriptome profiling of Nannochloropsis gaditana in nitrogen depletion. Molecular Plant, 7: 323-335.
Cha, T. S., Chen, C. F., Yee, W., Aziz, A., and Loh, S. H., 2011. Cinnamic acid, coumarin and vanillin: Alternative phenolic compounds for efficient Agrobacterium-mediated transformation of the unicellular green alga, Nannochloropsis sp. Journal of Microbiological Methods, 84: 430-434.
Chen, H. L., Li, S. S., Huang, R., and Tsai, H. J., 2008. Conditional production of a functional fish growth hormone in the transgenic line of Nannochloropsis oculata (Eustigmatophyceae). Journal of Phycology, 44: 768-776.
Crevillen, P., Yang, H., Cui, X., Greeff, C., Trick, M., Qiu, Q., Cao, X., and Dean, C., 2014. Epigenetic reprogramming that prevents transgenerational inheritance of the vernalized state. Nature, 515: 587-590.
Fawley, K. P., and Fawley, M. W., 2007. Observations on the diversity and ecology of freshwater Nannochloropsis (Eustigmatophyceae), with descriptions of new taxa. Protist, 158: 325-336.
Fedor, M. J., 2008. Alternative splicing minireview series: Combinatorial control facilitates splicing regulation of gene expression and enhances genome diversity. Journal of Biological Chemistry, 283 (3): 1209-1210.
Galloway, R. E., 1990. Selective condition and isolation of mutants in salt-tolerant, lipid-producing microalgae. Journal of Phycology, 26: 752-760.
Guillard, R. R., and Ryther, J. H., 1962. Studies of marine planktonic diatoms. I. Cyclotella nana Husted, and Detonula confervace (Cleve) Gran. Canadian Journal of Microbiology, 8 (2): 229-239.
Guillard, R. R. L., 1975. Culture of phytoplankton for feeding marine invertebrates. In: Culture of Marine Invertebrate Animals. Smith, W. L., and Chanley, M. H., eds., Plenum Press, New York, 26-60.
Hasegawa, P. M., Bressan, R. A., Zhu, J. K., and Bohnert, H. J., 2000. Plant cellular and molecular response to high salinity. Annual Review of Plant Physiology and Plant Molecular Biology, 51: 463-499.
Hoffmann, M., Marxen, K., Schulz, R., and Vanselow, K. H., 2010. TFA and EPA productivities of Nannochloropsis salina influenced by temperature and nitrate stimuli in turbidostatic controlled experiments. Marine Drugs, 8: 2526-2545.
Huang, A., He, L., and Wang, G., 2011. Identification and characterization of microRNAs from Phaeodactylum tricornutum by high-throughput sequencing and bioinformatics analysis.BMC Genomics, 12: 337.
Huang, W., and Hu, H., 2013. Studyon the salinity tolerance and oil accumulation in Nannochloropsis. Acta Hydrobiologica Sinica, 37 (2): 383-387 (in Chinese).
Kilian, O., Benemann, C. S., Niyogi, K. K., and Vick, B., 2011. High-efficiency homologous recombination in the oil-producing alga Nannochloropsis sp. Proceedings of the National Academy of Sciences of the United States of America, 108: 21265-21269.
Lempiainen, H., and Shore, D., 2009. Growth control and ribosome biogenesis. Current Opinion in Cell Biology, 21: 855-863.
Levinson, G., and Gutman, G. A., 1987. Slipped-strand mispairing: A major mechanism for DNA sequence evolution. Molecular Biology and Evolution, 4: 203-221.
Liang, C., Cao, S., Zhang, X., Zhu, B., Su, Z., Xu, D., Guang, X., and Ye, N., 2013. De novo sequencing and global transcriptome analysis of Nannochloropsis sp. (Eustigmatophyceae) following nitrogen starvation. Bioenergy Research, 6: 494-505.
Loewith, R., and Hall, M. N., 2011. Target of rapamycin (TOR) in nutrient signaling and growth control. Genetics, 189: 1177-1201.
Lohbeck, K. T., Riebesell, U., and Reusch, T. B. H., 2012. Adaptive evolution of a key phytoplankton species to ocean acidification. Nature Geoscience, 5: 346-351.
Ma, Y. B., Wang, Z. Y., Zhu, M., Yu, C. J., Cao, Y. P., Zhang, D. Y., and, Zhou, G. K., 2013. Increased lipid productivity and TAG content in Nannochloropsis by heavy ion irradiation mutagenesis. Bioresource Technology, 136: 360-367.
Mao, X., Cai, T., Olyarchuk, J. G., and Wei, L., 2005. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics, 21 (19): 3787-3793.
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L., and Wold, B., 2008. Mapping and quantifying mammalian transcriptomes by RNA-seq. Nature Methods, 5 (7): 621-628.
Pan, K. H., Qin, J. J., Li, S., Dai, W. K., Zhu, B. H., Jin, Y. C., Yu, W. G., and Yang, G. P., 2011. Nuclear monoploidy and asexual propagation of Nannochloropsis oceanica as revealed by its genome sequence. Journal of Phycology, 47: 1425-1432.
Radakovits, R., Jinkerson, R. E., Fuerstenberg, S. I., Tae, H., Settlage, R. E., Boore, J. L., and Posewitz, M. C., 2012. Draft genome sequence and genetic transformation of the oleaginous alga Nannochloropsis gaditana. Nature Communication, 3: 686.
Rodolfi, L., Chini Zittelli, G., Bassi, N., Padovani, G., Biondi, N., Bonini, G., and Tredici, M. R., 2009. Microalgae for oil: Strain selection, induction of lipid synthesis and outdoor mass cultivation in a low-cost photobioreactor. Biotechnology and Bioengineering, 102: 100-112.
Rodriguez-Navarro, A., and Rubio, F., 2006. High-affinity potassium and sodium transport systems in plants. Journal of Experimental Botany, 57 (5): 1149-1160.
Shi, J., Pan, K. H., Yu, J. Z., Zhu, B. H., Yang, G. P., Yu, W. G., and Zhang, X. Y., 2008. Analysis of the expressed sequence tags from the marine microalga Nannochloropsis oculata (Eustigmatophyceae). Journal of Phycology, 44: 99-102.
Stamm, S., Ben-Ari, S., Rafalska, I., Tang, Y., Zhang, Z., Toiber, D., Thanaraj, T. A., and Soreq, H., 2005. Function of alternative splicing. Gene, 344: 1-20.
Sukenik, A., Beardall, J., Kromkamp, J. C., Kopecky, J., Masojídek, J., Bergeijk, B., Gabai, G., and Shaham, E., 2009. Photosynthetic performance of outdoor Nannochloropsis mass cultures under a wide range of environmental conditions. Aquatic Microbial Ecology, 56: 297-308.
Sunday, J. M., Calosi, P., Dupont, S., Munday, P. L., Stillman, J. H., and Reusch, T. B. H., 2014. Evolution in an acidifying ocean. Trends in Ecology & Evolution, 29: 117-125.
Trapnell, C., Williams, B. A., Pertea, G., Mortazavi, A., Kwan, G., van Baren, M. J., Salzberg, S. L., Wold, B. J., and Pachter, L., 2010. Transcript assembly and abundance estimation from RNA-Seq reveals thousands of new transcripts and switching among isoforms. Nature Biotechnology, 28 (5): 511-515.
Trapnell, C., Roberts, A., Goff, L., Pertea, G., Kim, D., Kelley, D. R., Pimentel, H., Salzberg, S. L., Rinn, J., and Pachter, L., 2012. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature Protocols, 7 (3): 562-578.
Vieler, A., Wu, G., Tsai, C-H., Bullard, B., Cornish, A. J., Harvey, C., Reca, I-B., Thornburg, C., Achawanantakun, R., Buehl, C. J., Campbell, M. S., Cavalier, D., Childs, K. L., Clark, T. J., Deshpande, R., Erickson, E., Ferguson, A. N., Handee, W., Kong, Q., Li, X., Liu, B., Lundback, S., Peng, C., Roston, R. L., Sanjaya., Simpson, J. P., TerBush, A., Warakanont, J., Z?uner, S., Farre, E. M., Hegg, E. L., Jiang, N., Kuo, M-H., Lu, Y., Niyogi, K. K., Ohlrogge, J., Osteryoung, K. W., Shachar-Hill, Y., Sears, B. B., Sun, Y., Takahashi, H., Yandell, M., Shiu, S-H., and Benning, C., 2012. Genome, functional gene annotation, and nuclear transformation of the heterokont oleaginous alga Nannochloropsis oceanica CCMP1779. PLoS Genetics, 8 (11): e1003064.
Walker, D. J., Leigh, R. A., and Miller, A. J., 1996. Potassium homeostasis in vacuolate plant cells. Proceedings of the National Academy of Sciences of the United States of America, 93: 10510-10514.
Wang, D., Ning, K., Li, J., Hu, J., Han, D., Wang, H., Zeng, X., Jing, X., Zhou, Q., Su, X., Chang, X., Wang, A., Wang, W., Jia, J., Wei, L., Xin, Y., Qiao, Y., Huang, R., Chen, J., Han, B., Yoon, K., Hill, R. T., Zohar, Y., Chen, F., Hu, Q., and Xu, J., 2014. Nannochloropsis genomes reveal evolution of microalgal oleaginous traits. PLoS Genetics, 10 (1): e1004094.
Wang, Z., Gerstein, M., and Snyder, M., 2009. RNA-Seq: A revolutionary tool for transcriptomics. Nature Reviews Genetics, 10: 57-63.
Weeks, D. P., 2011. Homologous recombination in Nannochloropsis: A powerful tool in an industrially relevant alga. Proceedings of the National Academy of Sciences of the United States of America, 108: 20859-20860.
Young, M. D., Wakefield, M. J., Smyth, G. K., and Oshlack, A., 2010. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biology, 11: R14.
(Edited by Qiu Yantao)
(Received June 14, 2014; revised November 26, 2014; accepted March 26, 2015)
J. Ocean Univ. China (Oceanic and Coastal Sea Research)
DOI 10.1007/s11802-015-2689-7
ISSN 1672-5182, 2015 14 (5): 922-930
http://www.ouc.edu.cn/xbywb/
E-mail:xbywb@ouc.edu.cn
* Corresponding author. Tel: 0086-532-82031636
E-mail: yguanpin@mail.ouc.edu.cn
 Journal of Ocean University of China2015年5期
Journal of Ocean University of China2015年5期
- Journal of Ocean University of China的其它文章
- A Carboxymethyl Cellulase from a Marine Yeast (Aureobasidium pullulans 98): Its Purification, Characterization, Gene Cloning and Carboxymethyl Cellulose Digestion
- Effects of Dietary Stachyose on Growth Performance, Digestive Enzyme Activities and Intestinal Morphology of Juvenile Turbot (Scophthalmus maximus L)
- Pharmacokinetics and Biodegradation of Chitosan in Rats
- Pharmacokinetics and Biodegradation Performance of a Hydroxypropyl Chitosan Derivative
- Changes in Plasma Osmolality, Cortisol and Amino Acid Levels of Tongue Sole (Cynoglossus semilaevis) at Different Salinities
- Effects of Different Microbes on Fermenting Feed for Sea Cucumber (Apostichopus japonicus)