
Accelerated evolution of constraint elements for hematophagic adaptation in mosquitoes
2015-04-10 00:58:33MingShanWANGAdeniyiADEOLAYanLIYaPingZHANGDongDongWU
Zoological Research 2015年6期
Ming-Shan WANG, Adeniyi C. ADEOLA, Yan LI, Ya-Ping ZHANG,3,*, Dong-Dong WU,*
1State Key Laboratory of Genetic Resources and Evolution, Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming 650223, China
2University of Chinese Academy of Sciences, Beijing 100049, China
3 Laboratory for Conservation and Utilization of Bio-resources, Yunnan University, Kunming 650091, China
INTRODUCTION
Recent advances in DNA sequencing technology and comparative genomic analysis have unlocked many mysteries of genetic variation in phenotype determination and have greatly changed the landscape of biological research (Chen,2015; Kim et al, 2011; Li et al, 2010). With the application of next-generation sequencing (NGS), whole genome sequencing of organisms of interest has become less time and cost consumptive. Draft genomes of many non-model species have been completed in the past five years. Comparative analysis of related whole genomes is a useful approach in genome interpretation (Chen, 2015; Zeng et al, 2013). This has facilitated the discovery of functionally important genomic elements responsible for specific phenotypes and identification of adaptive genomic imprints that occurred during evolution(Lindblad-Toh et al, 2011).1
Mosquitoes are found within the dipteran flies (Culicidae),and feed on vertebrate blood (such as human, bird, rat and livestock). Many mosquito-borne pathogens, such as malaria,lymphatic ベlariasis and dengue fever, can be transmitted between humans and other vertebrates during blood feeding,causing a large number of deaths worldwide annually,particularly in Africa (Tsuji et al, 1990a). This demonstrates the extent of the threat on global health imposed by mosquitoes.Blood nutrients such as proteins and iron are essential for female mosquitoes to lay eggs and complete the reproductive process, even though there are variations in reproductive strategies between autogenous and anautogenous mosquitoes(Tsuji et al, 1990b). Mosquitoes are capable of ingesting a large volume of blood in excess of their body weight during a single meal (Friend et al, 1965), exposing them to high temperature and osmotic pressure and oxygen toxicity from heme, iron and ROS, which came from the processes of blood digestion and mitochondrial metabolism.
In addition, due to their long divergence time and various habitats, different mosquitoes have evolved variation in phenotype, feeding behavior and host preference. A good understanding of the genome components and their role in mosquito species[0] may help determine the mechanisms of specific traits (i.e., hematophagy), and provide a potential starting point to ascertain the driving forces of disease transmission and develop measures to effectively control mosquito-borne diseases.
Constraint elements (CEs) are a group of genomic regions that evolved under restriction across different species over a long period of evolutionary time, and have been widely studied in vertebrate, insect, worm and yeast genomes (Lindblad-Toh et al, 2011; Siepel et al, 2005; Tian et al, 2009; Woolfe et al, 2005).Constraint in a genome sequence implies that it is conserved and plays an important role in biological function. Recent and rapid evolution in a group of CEs in a lineage (hereafter called accelerated elements (AEs)) implies that adaptive evolution may have occurred (Amemiya et al, 2013; Holloway et al, 2008;Lindblad-Toh et al, 2011). Comprehensive investigation of CEs and AEs at the genomic-wide scale has emerged as a useful research tool in evolutionary study and has provided directions to test hypotheses and design experiments (Amemiya et al,2013; Lindblad-Toh et al, 2011). With the development of whole genome sequencing technologies, the genomes of four bloodfeeding insects (Aedes aegypti(Aa) (Nene et al, 2007),Anopheles darling (Ad) (Marinotti et al, 2013), Anophelesgambia(Ag) (Holt et al, 2002) andCulex quinquefasciatus(Cq)(Arensburger et al, 2010), along with 12 previously published(Clark et al, 2007) Drosophila genomes (Drosophilamelanogaster, D. pseudoobscura, D. ananassae, D. erecta, D.grimshawi, D. mojavensis, D. persimilis, D. sechellia, D.simulans, D. virilis, D. willistoni,andD. yakuba) were retrieved from the UCSC Genome Brower and the Ensembl Metazoa database (version 20). Whole genome comparative analysis of these genomes provides the opportunity to comprehensively interpret the genomic evolution of Diptera and the molecular mechanism of hematophagy adaptation in mosquitoes. We carried out a comprehensive search for CEs through genomewide multiple alignments of the Diptera genomes (four mosquitoes and 12 Drosophilae) to predict the AE divergence of constraint genomic regions in specific mosquito lineages.
MATERIALS AND METHODS
Whole genome alignments
Genomes of 12 Drosophila, including D. melanogaster (dm3), D.pseudoobscura (dp4), D. ananassae (droAna3), D. erecta(droEre2), D. grimshawi (droGri2), D. mojavensis (droMoj3), D.persimilis (droPer1), D. sechellia (droSec1), D. simulans(droSim1), D. virilis (droVir3), D. willistoni (droWil1), and D.yakuba (droYak2), and four mosquito species (Aedes aegypti,Anophelesdarling,AnophelesgambiaandCulexquinquefasciatus) were download from the UCSC Genome Brower and Ensembl Metazoa database (version 20). With the exception of Aedes aegypti, Anopheles darling, Anophelesgambia, Culex quinquefasciatusandD. melanogaster,reference base pair wise alignments for all species were download from the UCSC Genome Brower. Using the D.melanogaster(BDGP assembly release 5) genome as a reference, pair wise alignments were performed using the LASTZ (http://www.bx.psu.edu/miller_lab/) program with parameters set to “E=30 O=400 K=3000 L=2200 M=50 --format=axt”. To reduce single coverage with respect to the reference genome and obtain long-linked and longer syntenic contiguous alignments, the alignment blocks were passed through “chaining” and “netting” processes using axtChain,ChainNet and netSyntenic from UCSC tools, as described previously (Kent et al, 2003). SixteenD. melanogaster-centric whole genome alignments were generated using MULTIZ(Blanchette et al, 2004) v012109/roast and graded by the phylogenic topology tree in Figure 1A.
Evolutionary constraint elements detection
To measure conservation tracks of alignment, four-fold degenerate sites were extracted from chromosomes (2R, 2L,3R, 3L) of MUTIZ alignments using msa_view software from the PHAST (Siepel et al, 2005) package based on a gff format gene annotation file, and a neutral model (non-conserved model) was then generated using PhyloFit. Conservation scores and conserved sequences were predicted using PhastCons.Running parameters were set with an average conserved sequence length of 45 bp (“--expected-length 45”), target coverage of input alignments was set to 0.3 (“--target-coverage 0.3”), and scaling factor for non-conserved model was set to 0.3(“--rho 0.3”). Elements with length <30 bp were discarded. The non-conserved phylogenies were estimated by PhyloFit using four-fold degenerate sites, as shown in Figure 1A.
Lineage-specific accelerated elements identification
Lineage-specific accelerated elements were identified by first defining candidate genomic regions across all Diptera,excluding the lineage of interest. Conserved regions were identified using the PhastCons program, as mentioned above,and the pipeline parameters were set to “--expected-length 45 --target-coverage 0.3 --rho 0.3”.[0] Lineage-specific accelerated elements were identified by likelihood ratio tests (Holloway et al,2008) implemented using PhyloP software from the PHAST package by comparing conserved region substitution rate scores between lineages of interest and the rest of the subtree.A neutral model for inference was created by the above mentioned methods using PhyloFit. PhyloP was run with the scoring system set to “CONACC”. Lineages used in accelerated element detection (lineage of interest) included Aedes aegypti,Anopheles darling, Anopheles gambia, Culex quinquefasciatusand the common ancestor of mosquitoes (RCAM). The element with “alt_subscale >1” showed the region that evolved faster than the remaining part of the tree, thereby providing evidence of acceleration. AP-value of <0.01 was used as the cutoff(FDR-adjusted).
Element genomic location and functional category enriched analysis
All CEs and AEs were annotated withD. melanogasterENSEMBLE gene annotations using ANNOVAR (Wang et al,2010), and the genomic regions were classified as down- and up-stream, exonic, intergenic, untranslated and intronic regions.Functional enrichment and pathway annotation clustering were performed using the DAVID (Database for Annotation Visualization and Integrated Discovery) tool (Huang et al, 2008).

Figure 1 Distribution of constraint elements in 16 Diptera genomes
RESULTS
Evolutionary constraint elements (CEs) in Diptera
Genome-wide CEs in Diptera (four mosquitoes and 12Drosophila) were retrieved using PhastCons (see Materials and Methods). A total of 547 726 CEs with length ranging from 30 bp to 6 500 bp (mean size 125.6 bp) were identified (Figure 1B),covering 40.8% of the D. melanogaster genome. About 38% of CEs were located in the protein-coding gene regions, which included 23.10% in the coding region, 8.27% in the untranslated regions (3′UTR and 5′UTRs) and 6.80% within 1 kb of the upstream and downstream regions. A large proportion of CEs were found in the intronic and intergenic regions(39.30% and 21.47%, respectively) (Figure 1C). The distribution pattern in the dipteran genome was similar to that of vertebrate genomes, indicating the potentially important role of non-coding regions in both vertebrates and dipterans. However, the proportion of the dipteran genome covered by CEs was higher than that in mammals (~4.5% of mammal genome) (Lindblad-Toh et al, 2011), which may relate to the different properties of the insect genome compared with the mammal genome, such as higher gene density and more compact gene distribution(Bird et al, 2007; Holloway et al, 2008; Lindblad-Toh et al, 2011).The constraint element results in our study were in accordance with previous identification in the four insects (Anophelesgambia, D. melanogaster, D. yakubaandD. pseudoobscura),except for a slight difference in the proportion of constraint element composition, which may be due to an increased number of higher quality genome sequences used in whole genome alignment (Siepel et al, 2005).
Accelerated conserved elements in mosquitoes Identification of lineage-specific accelerated elements by likelihood ratio tests
We carried out a series of likelihood ratio tests to retrieve elements showing rapid evolution of the constraint regions in each mosquito lineage using PhyloP software. Constraint genomic regions used for acceleration detection were obtained using the same method as described above by excluding the lineage of interest across all 16 dipteran genome alignments(see Materials and Methods). We retrieved elements showing a divergence of constraint regions in RCAM, Aa, Ad, Ag and Cq lineages, respectively.
In total, 1 463, 199, 954, 660 and 779 AEs in RCAM, Aa, Ad,Ag and Cq lineages were identified, respectively (Figure 2A).These AEs showed a similar length distribution pattern (Figure 2B). The Phylop scores for AEs were negatively correlated with their lengths (P<0.01) (Figure 2C). The majority of AEs were assigned to the protein-coding genomic region, but a few were found in the non-coding region (Figure 2D). This distribution pattern was similar to the AEs identified in D. melanogaster, but different to human and primate lineages with a large number of elements in the non-coding regions (Lindblad-Toh et al, 2011).These findings may be the result of the higher density and compactness of gene distribution in insect genomes compared with that of vertebrate genomes (Bird et al, 2007; Holloway et al,2008; Lindblad-Toh et al, 2011).
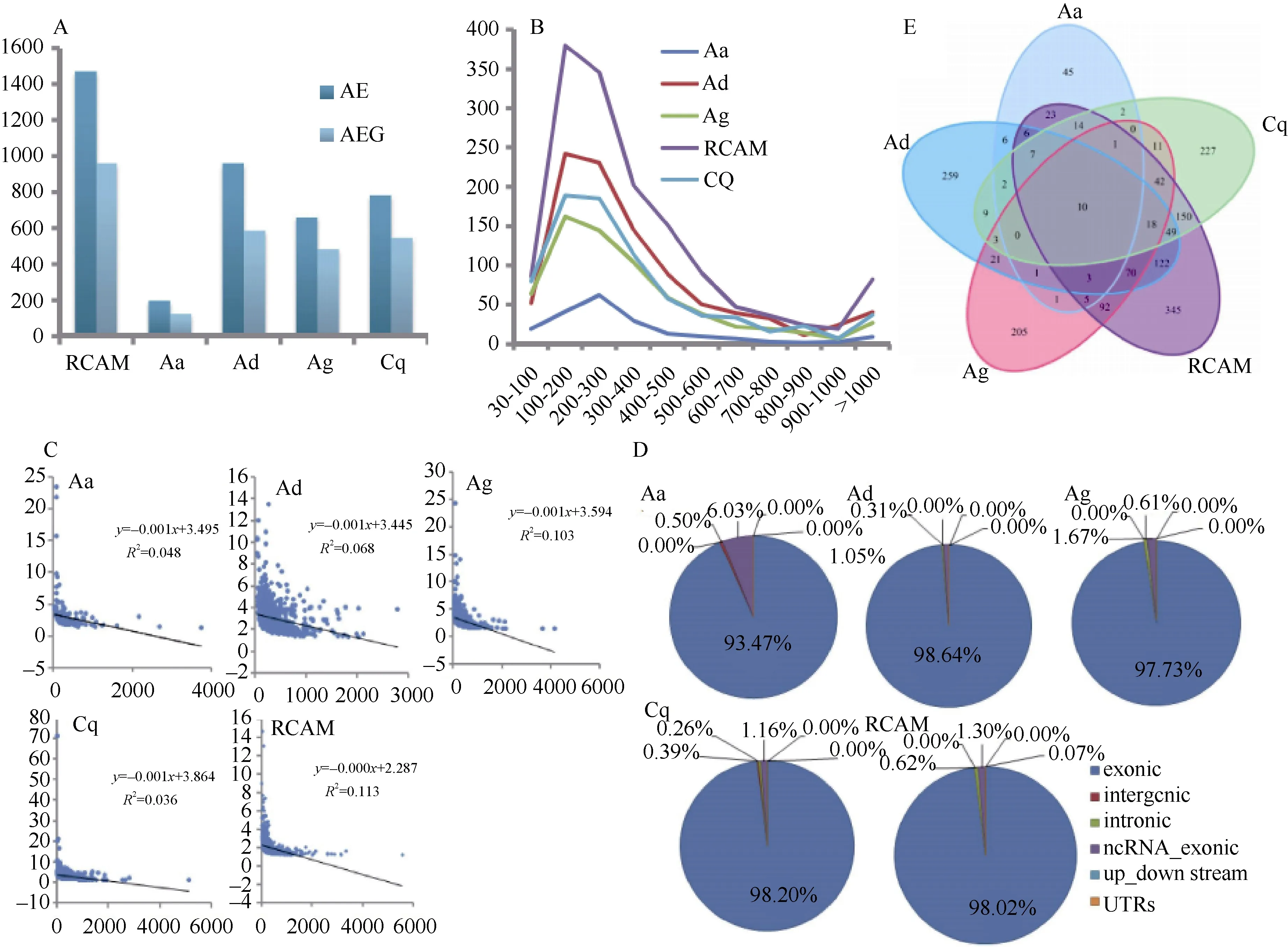
Figure 2 Landscape of CEs, AEs and AEGs detected in mosquitoes
Functional enrichment analysis of AEs
The AEs were annotated with D. melanogaster ENSEMBLE gene annotations using ANNOVAR (Wang et al, 2010), and functional enrichment analysis of protein-codon genes covered by AEs (here after referred to as AEGs) were performed using DAVID, including KEGG pathways and GO categories. We found that these AEGs were enriched in a wide variety of functional categories in different lineages. In total, 957, 126, 586,483 and 545 AGEs were identified in RCAM, Aa, Ad, Ag and Cq lineages, respectively (Figure 2A). Of these, only 10 AEGs were shared by RCAM and the other lineages (Figure 2E).
We found that AEGs in RCAM showed divergence from CEs,and these AEGs were enriched in various functional categories.However, some were over-presented in metabolic-related GO categories, including iron transport/binding, metal ion transport/binding, potassium ion transport/binding, lipid metabolic process, iron, metal ion and potassium ion channel activity (P<0.05) (Supplementary Table 1). This finding was not unexpected because compared withDrosophila, mosquitoes have the ability to handle high levels of iron, lipids and heme after ingesting a large volume of blood (Graca-Souza et al,2006). In addition, some genes participated in exopeptidase,metalloexopeptidase, metallocarboxypeptidase, metallopeptidase and hydrolase activities (P<0.05) (Supplementary Table 1). For instance, exopeptidases, one of the two major classes of secreted proteases in the midgut, function as carboxypeptidases,while amino peptides degrade polypeptides from the terminal ends into exopeptides (Isoe et al, 2009). Therefore, the rapid evolution of these genes in mosquitoes may explain their high efficiency in blood feeding and digestion. Imbibing a large volume of blood from a warm-blooded, vertebrate host not only quickly generates osmotic stress, which requires an efficient excretory system for rapid removal of excess water, but also increases the mosquito′s body temperature as blood enters the gut (Benoit et al, 2011). Interestingly, we found heat shock protein 26 (Hsp26) with rapid evolution in RCAM, which has been reported with increased expression when mosquitoes are exposed to high temperature (42°C) (Zhao et al, 2010).
Due to the long-term and constant use of insecticides,mosquitoes have shown partial tolerance or resistance to insecticides such as DTT, permethrin and malathion. Certain genes or mutations may have contributed to this drug tolerance or resistance in mosquitoes (You et al, 2013). As expected, we found many genes showing evolutionary acceleration enrichment in detoxification and drug metabolism GO categories and pathways, such as response to toxic substances,response to arsenic-containing substances, and detoxification of arsenic-containing substances (P<0.05) (Supplementary Table 1). Three genes,GSTD1, GSTS1andGSTD9, belong to the glutathione s-transferase (GST) family of proteins, which are involved in the detoxification of a wide range of xenobiotics and protection from oxidative damage, as well as the intracellular transport of hormones, endogenous metabolites and exogenous chemicals including insecticides [0](Sanil et al,2014). Our findings also revealed that CYP4C3, CYP4D8 andCYP4P1, members of the cytochrome P450 monooxygenases(CYPs), showed rapid evolution. These genes play vital roles in insecticide resistance and enhance higher rates of insecticide metabolism (Chandor-Proust et al, 2013).
Via analysis of the AEGs detected in the studied lineages, we discovered many genes showed rapid evolution, which were functionally associated with various ion, ATP, lipid and glucose metabolic processes in Aa, Ad, Ag and Cq (Supplementary Table 2-5), and with hydrolase activity in Ad, Ag and [0]Cq(Supplementary Table 3-5). In Cq, more genes and GO categories related to blood feeding showed rapid evolution compared with the other lineages (Figure 2A), including ion,ATP, lipid hydrolase activity and glucose metabolism categories,exopeptidase, metalloexopeptidase, metallocarboxypeptidase and metallopeptidase activities, but few blood feeding related genes and terms were found in Aa, Ad and Ag (Supplementary Table 2-5). More genes showing CE acceleration in Cq may help explain how these diverse blood feeders effectively and successfully digest blood from multiple hosts, including birds,humans and livestock. In Ag, Ad and Cq, we also found several genes enriched in the functional categories of xenobiotic transporter activity, drug transporter activity and xenobiotictransporting ATPase activity, and most were members of CYPs and GSTs, such as GSTD4, GSTD9, CYP4D8, and CYP6A9(Supplementary Table 3-5).
DISCUSSION
We identified more than 540 000 genomic regions (>=30 bp)that evolved under constraint over several million years in Diptera flies by comparative analysis of multiple alignments of 12 Drosophilae and four mosquito genomes. Most CEs were categorized into intergenic, intronic and exonic regions, similar in proportion with that of vertebrates and previous reports(Holloway et al, 2008; Siepel et al, 2005). A higher fraction of CEs (covering 40.8% of the D. melanogaster genome) were identified in the dipteran genome than that identified in mammals (~4.5% of the mammal genome) (Lindblad-Toh et al,2011), which may be due to the higher density and compactness of gene distribution in the insect genome compared with that of the vertebrate genome (Bird et al, 2007;Holloway et al, 2008; Lindblad-Toh et al, 2011).
Being conserved in a genome sequence implies constraint in function. Genomic elements in a lineage that have been under constraint over long periods of evolutionary time, but which have experienced recent and rapid evolutionary expansion,suggest adaptive evolution in those lineages (Holloway et al,2008). We identified AEs in RCAM, Aa, Ad, Ag and Cq lineages,respectively. Contrary to CEs, the majority of AEs were found in the protein-coding genomic region, with a small proportion located in the non-coding region. This distribution pattern is similar to the AEs identified in the D. melanogaster lineage reported previously (Holloway et al, 2008), but different from human and primate lineages in which a large number of elements are assigned into the non-coding regions (Lindblad-Toh et al, 2011). Due to the larger genome size and general biological complexity of vertebrates compared with insects,greater fractions of AEs are found in the non-coding genomic regions in vertebrates. This is likely a reflection of the important role of other non-coding sequences (i.e., lncRNA and circ-RNA)in eukaryotes, which serve as important regulatory factors to regulate gene expression (Lindblad-Toh et al, 2011; Siepel et al,2005), as verified in recent studies (Iyer et al, 2015; Zhang et al,2013).
Compared with Drosophila, blood feeding is an intrinsic characteristic of mosquitoes and nutrients in blood are essential for completing reproductive processes. Mosquitoes suffer osmotic and temperature stress after ingesting a large volume of blood, but can deal with it effectively. Comparison of the mosquito genome with that of Drosophila may provide insight into the factors responsible for the above traits. As expected,we discovered some genes related to the transportation and binding of iron, lipids and heme and the digestion of blood essential exopeptidase. We also found hydrolase-related genes,which showed divergence from the CEs in the mosquito lineages and which may contribute to the evolution of blood feeding habits after the divergence from Drosophilae.[0] In addition, some of the detoxification and insecticide resistancerelated genes discovered (i.e., glutathione s-transferases (GSTs)and cytochrome P450s) showed rapid evolution in mosquitoes,which may be an adaptive change due to the constant use of chemical insecticides. Genes related to blood feeding adaptation and insecticide resistance also showed rapid evolution in RCAM. While this further evolved in certain mosquitoes (Ag, Aa, Ad and Cq lineages), few genes were shared by RCAM and the other lineages (Figure 2E). This implies that mosquitoes probably possess an independent mechanism and/or different ability to adapt to blood meals or drug resistance. Earlier evidence showed that mosquitoes exhibit differences in host preference (Takken & Verhulst, 2013).For instance, Cq is a diverse blood feeder that can effectively and successfully digest blood from multiple hosts, including birds, humans and livestock. In Cq, we discovered more genes that displayed rapid evolution (Figure 2) and more GO categories related to blood feeding adaptation than any other lineage. This finding indicates that although the same biological systems were universally fast-evolving, adaptations occurred through somewhat distinct degrees of evolution in different mosquito clades (Lindblad-Toh et al, 2011).
Our study had a number of limitations. First, some of the genomes used in our comparison are recently drafted genomes with potential sequencing and assembly errors, which may lead to bias. In addition, multiple-species, whole genome comparisons are a complex and tedious task, and might led to alignment errors (incorrect alignment blocks or alignment failures) resulting from the program used. Conserved elements and lineage-specific elements estimated here were more or less constrained and fast-evolved than in reality. Our analysis focused on AEGs and did not assign a gene or mutation to specific traits, while recent studies have shown that lineagespecific accelerated elements (LAEs) have important roles in biological processes (Iyer et al, 2015). Improving genome accuracy, alignment quality and functional characterization of accelerated elements in mosquitoes, including LAEs, are important for future study, and may improve understanding of the biological, especially lineage-specific traits of mosquitoes.
Amemiya CT, Alfoldi J, Lee AP, Fan S, Philippe H, Maccallum I, Braasch I,Manousaki T, Schneider I, Rohner N, Organ C, Chalopin D, Smith JJ,Robinson M, Dorrington RA, Gerdol M, Aken B, Biscotti MA, Barucca M,Baurain D, Berlin AM, Blatch GL, Buonocore F, Burmester T, Campbell MS,Canapa A, Cannon JP, Christoffels A, De Moro G, Edkins AL, Fan L, Fausto AM, Feiner N, Forconi M, Gamieldien J, Gnerre S, Gnirke A, Goldstone JV,Haerty W, Hahn ME, Hesse U, Hoffmann S, Johnson J, Karchner SI,Kuraku S, Lara M, Levin JZ, Litman GW, Mauceli E, Miyake T, Mueller MG,Nelson DR, Nitsche A, Olmo E, Ota T, Pallavicini A, Panji S, Picone B,Ponting CP, Prohaska SJ, Przybylski D, Saha NR, Ravi V, Ribeiro FJ,Sauka-Spengler T, Scapigliati G, Searle SM, Sharpe T, Simakov O, Stadler PF, Stegeman JJ, Sumiyama K, Tabbaa D, Tafer H, Turner-Maier J, Van Heusden P, White S, Williams L, Yandell M, Brinkmann H, Volff JN, Tabin CJ, Shubin N, Schartl M, Jaffe DB, Postlethwait JH, Venkatesh B, Di Palma F, Lander ES, Meyer A, Lindblad-Toh K. 2013. The African coelacanth genome provides insights into tetrapod evolution.Nature, 496(7445): 311-316.
Arensburger P, Megy K, Waterhouse RM, Abrudan J, Amedeo P, Antelo B,Bartholomay L, Bidwell S, Caler E, Camara F, Campbell CL, Campbell KS,Casola C, Castro MT, Chandramouliswaran I, Chapman SB, Christley S,Costas J, Eisenstadt E, Feschotte C, Fraser-Liggett C, Guigo R, Haas B,Hammond M, Hansson BS, Hemingway J, Hill SR, Howarth C, Ignell R,Kennedy RC, Kodira CD, Lobo NF, Mao C, Mayhew G, Michel K, Mori A,Liu N, Naveira H, Nene V, Nguyen N, Pearson MD, Pritham EJ, Puiu D, Qi Y, Ranson H, Ribeiro JMC, Roberston HM, Severson DW, Shumway M,Stanke M, Strausberg RL, Sun C, Sutton G, Tu Z, Tubio JMC, Unger MF,Vanlandingham DL, Vilella AJ, White O, White JR, Wondji CS, Wortman J,Zdobnov EM, Birren B, Christensen BM, Collins FH, Cornel A, Dimopoulos G, Hannick LI, Higgs S, Lanzaro GC, Lawson D, Lee NH, Muskavitch MaT,Raikhel AS, Atkinson PW. 2010. Sequencing ofCulex quinquefasciatusestablishes a platform for mosquito comparative genomics.Science,330(6000): 86-88.
Benoit JB, Lopez-Martinez G, Patrick KR, Phillips ZP, Krause TB, Denlinger DL. 2011. Drinking a hot blood meal elicits a protective heat shock response in mosquitoes.Proceedings of the National Academy of Sciences of the United States of America, 108(19): 8026-8029.
Bird CP, Stranger BE, Liu M, Thomas DJ, Ingle CE, Beazley C, Miller W,Hurles ME, Dermitzakis ET. 2007. Fast-evolving noncoding sequences in the human genome.Genome Biology, 8(6): R118.
Blanchette M, Kent WJ, Riemer C, Elnitski L, Smit AF, Roskin KM, Baertsch R, Rosenbloom K, Clawson H, Green ED, Haussler D, Miller W. 2004.Aligning multiple genomic sequences with the threaded blockset aligner.Genome Research, 14(4): 708-715.
Chandor-Proust A, Bibby J, Regent-Kloeckner M, Roux J, Guittard-Crilat E,Poupardin R, Riaz MA, Paine M, Dauphin-Villemant C, Reynaud S, David JP. 2013. The central role of mosquito cytochrome P450 CYP6Zs in insecticide detoxification revealed by functional expression and structural modelling.The Biochemical Journal, 455(1): 75-85.
Chen H. 2015. Population genetic studies in the genomic sequencing era.Zoological Research, 36(4): 223-232.
Clark AG, Eisen MB, Smith DR, Bergman CM, Oliver B, Markow TA,Kaufman TC, Kellis M, Gelbart W, Iyer VN, Pollard DA, Sackton TB,Larracuente AM, Singh ND, Abad JP, Abt DN, Adryan B, Aguade M, Akashi H, Anderson WW, Aquadro CF, Ardell DH, Arguello R, Artieri CG, Barbash DA, Barker D, Barsanti P, Batterham P, Batzoglou S, Begun D, Bhutkar A,Blanco E, Bosak SA, Bradley RK, Brand AD, Brent MR, Brooks AN, Brown RH, Butlin RK, Caggese C, Calvi BR, Bernardo de Carvalho A, Caspi A,Castrezana S, Celniker SE, Chang JL, Chapple C, Chatterji S, Chinwalla A,Civetta A, Clifton SW, Comeron JM, Costello JC, Coyne JA, Daub J, David RG, Delcher AL, Delehaunty K, Do CB, Ebling H, Edwards K, Eickbush T,Evans JD, Filipski A, Findeiss S, Freyhult E, Fulton L, Fulton R, Garcia AC,Gardiner A, Garfield DA, Garvin BE, Gibson G, Gilbert D, Gnerre S,Godfrey J, Good R, Gotea V, Gravely B, Greenberg AJ, Griffiths-Jones S,Gross S, Guigo R, Gustafson EA, Haerty W, Hahn MW, Halligan DL,Halpern AL, Halter GM, Han MV, Heger A, Hillier L, Hinrichs AS, Holmes I,Hoskins RA, Hubisz MJ, Hultmark D, Huntley MA, Jaffe DB, Jagadeeshan S, Jeck WR, Johnson J, Jones CD, Jordan WC, Karpen GH, Kataoka E,Keightley PD, Kheradpour P, Kirkness EF, Koerich LB, Kristiansen K,Kudrna D, Kulathinal RJ, Kumar S, Kwok R, Lander E, Langley CH, Lapoint R, Lazzaro BP, Lee SJ, Levesque L, Li R, Lin CF, Lin MF, Lindblad-Toh K,Llopart A, Long M, Low L, Lozovsky E, Lu J, Luo M, Machado CA,Makalowski W, Marzo M, Matsuda M, Matzkin L, McAllister B, McBride CS,McKernan B, McKernan K, Mendez-Lago M, Minx P, Mollenhauer MU,Montooth K, Mount SM, Mu X, Myers E, Negre B, Newfeld S, Nielsen R,Noor MA, O′Grady P, Pachter L, Papaceit M, Parisi MJ, Parisi M, Parts L,Pedersen JS, Pesole G, Phillippy AM, Ponting CP, Pop M, Porcelli D,Powell JR, Prohaska S, Pruitt K, Puig M, Quesneville H, Ram KR, Rand D,Rasmussen MD, Reed LK, Reenan R, Reily A, Remington KA, Rieger TT,Ritchie MG, Robin C, Rogers YH, Rohde C, Rozas J, Rubenfield MJ, Ruiz A, Russo S, Salzberg SL, Sanchez-Gracia A, Saranga DJ, Sato H,Schaeffer SW, Schatz MC, Schlenke T, Schwartz R, Segarra C, Singh RS,Sirot L, Sirota M, Sisneros NB, Smith CD, Smith TF, Spieth J, Stage DE,Stark A, Stephan W, Strausberg RL, Strempel S, Sturgill D, Sutton G, Sutton GG, Tao W, Teichmann S, Tobari YN, Tomimura Y, Tsolas JM, Valente VL,Venter E, Venter JC, Vicario S, Vieira FG, Vilella AJ, Villasante A, Walenz B,Wang J, Wasserman M, Watts T, Wilson D, Wilson RK, Wing RA, Wolfner MF, Wong A, Wong GK, Wu CI, Wu G, Yamamoto D, Yang HP, Yang SP,Yorke JA, Yoshida K, Zdobnov E, Zhang P, Zhang Y, Zimin AV, Baldwin J,Abdouelleil A, Abdulkadir J, Abebe A, Abera B, Abreu J, Acer SC, Aftuck L,Alexander A, An P, Anderson E, Anderson S, Arachi H, Azer M,Bachantsang P, Barry A, Bayul T, Berlin A, Bessette D, Bloom T, Blye J,Boguslavskiy L, Bonnet C, Boukhgalter B, Bourzgui I, Brown A, Cahill P,Channer S, Cheshatsang Y, Chuda L, Citroen M, Collymore A, Cooke P,Costello M, D′Aco K, Daza R, De Haan G, DeGray S, DeMaso C, Dhargay N, Dooley K, Dooley E, Doricent M, Dorje P, Dorjee K, Dupes A, Elong R,Falk J, Farina A, Faro S, Ferguson D, Fisher S, Foley CD, Franke A,Friedrich D, Gadbois L, Gearin G, Gearin CR, Giannoukos G, Goode T,Graham J, Grandbois E, Grewal S, Gyaltsen K, Hafez N, Hagos B, Hall J,Henson C, Hollinger A, Honan T, Huard MD, Hughes L, Hurhula B, Husby ME, Kamat A, Kanga B, Kashin S, Khazanovich D, Kisner P, Lance K, Lara M, Lee W, Lennon N, Letendre F, LeVine R, Lipovsky A, Liu X, Liu J, Liu S,Lokyitsang T, Lokyitsang Y, Lubonja R, Lui A, MacDonald P, Magnisalis V,Maru K, Matthews C, McCusker W, McDonough S, Mehta T, Meldrim J,Meneus L, Mihai O, Mihalev A, Mihova T, Mittelman R, Mlenga V,Montmayeur A, Mulrain L, Navidi A, Naylor J, Negash T, Nguyen T, Nguyen N, Nicol R, Norbu C, Norbu N, Novod N, O′N(xiāo)eill B, Osman S, Markiewicz E,Oyono OL, Patti C, Phunkhang P, Pierre F, Priest M, Raghuraman S, Rege F, Reyes R, Rise C, Rogov P, Ross K, Ryan E, Settipalli S, Shea T, Sherpa N, Shi L, Shih D, Sparrow T, Spaulding J, Stalker J, Stange-Thomann N,Stavropoulos S, Stone C, Strader C, Tesfaye S, Thomson T, Thoulutsang Y,Thoulutsang D, Topham K, Topping I, Tsamla T, Vassiliev H, Vo A,Wangchuk T, Wangdi T, Weiand M, Wilkinson J, Wilson A, Yadav S, Young G, Yu Q, Zembek L, Zhong D, Zimmer A, Zwirko Z, Jaffe DB, Alvarez P,Brockman W, Butler J, Chin C, Gnerre S, Grabherr M, Kleber M, Mauceli E,MacCallum I. 2007. Evolution of genes and genomes on the Drosophila phylogeny.Nature, 450(7167): 203-218.
Friend WG, Choy CT, Cartwright E. 1965. The effect of nutrient intake on the development and the egg production of Rhodnius prolixus Stahl(Hemiptera: Reduviidae).Canadian Journal of Zoology, 43(6): 891-904.
Graca-Souza AV, Maya-Monteiro C, Paiva-Silva GO, Braz GR, Paes MC,Sorgine MH, Oliveira MF, Oliveira PL. 2006. Adaptations against heme toxicity in blood-feeding arthropods.Insect Biochemistry and Molecular Biology, 36(4): 322-335.
Holloway AK, Begun DJ, Siepel A, Pollard KS. 2008. Accelerated sequence divergence of conserved genomic elements in Drosophila melanogaster.Genome Research, 18(10): 1592-1601.
Holt RA, Subramanian GM, Halpern A, Sutton GG, Charlab R, Nusskern DR, Wincker P, Clark AG, Ribeiro JM, Wides R, Salzberg SL, Loftus B,Yandell M, Majoros WH, Rusch DB, Lai Z, Kraft CL, Abril JF, Anthouard V,Arensburger P, Atkinson PW, Baden H, de Berardinis V, Baldwin D, Benes V, Biedler J, Blass C, Bolanos R, Boscus D, Barnstead M, Cai S, Center A,Chaturverdi K, Christophides GK, Chrystal MA, Clamp M, Cravchik A,Curwen V, Dana A, Delcher A, Dew I, Evans CA, Flanigan M,Grundschober-Freimoser A, Friedli L, Gu Z, Guan P, Guigo R, Hillenmeyer ME, Hladun SL, Hogan JR, Hong YS, Hoover J, Jaillon O, Ke Z, Kodira C,Kokoza E, Koutsos A, Letunic I, Levitsky A, Liang Y, Lin JJ, Lobo NF, Lopez JR, Malek JA, McIntosh TC, Meister S, Miller J, Mobarry C, Mongin E,Murphy SD, O′Brochta DA, Pfannkoch C, Qi R, Regier MA, Remington K,Shao H, Sharakhova MV, Sitter CD, Shetty J, Smith TJ, Strong R, Sun J,Thomasova D, Ton LQ, Topalis P, Tu Z, Unger MF, Walenz B, Wang A,Wang J, Wang M, Wang X, Woodford KJ, Wortman JR, Wu M, Yao A,Zdobnov EM, Zhang H, Zhao Q, Zhao S, Zhu SC, Zhimulev I, Coluzzi M,della Torre A, Roth CW, Louis C, Kalush F, Mural RJ, Myers EW, Adams MD, Smith HO, Broder S, Gardner MJ, Fraser CM, Birney E, Bork P, Brey PT, Venter JC, Weissenbach J, Kafatos FC, Collins FH, Hoffman SL. 2002.The Genome Sequence of the Malaria Mosquito Anopheles gambiae.Science, 298(5591): 129-149.
Huang DW, Sherman BT, Lempicki RA. 2008. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources.Nature Protocols, 4(1): 44-57.
Isoe J, Rascon AA, Jr., Kunz S, Miesfeld RL. 2009. Molecular genetic analysis of midgut serine proteases in Aedes aegypti mosquitoes.Insect Biochemistry and Molecular Biology, 39(12): 903-912.
Iyer MK, Niknafs YS, Malik R, Singhal U, Sahu A, Hosono Y, Barrette TR,Prensner JR, Evans JR, Zhao S, Poliakov A, Cao X, Dhanasekaran SM,Wu YM, Robinson DR, Beer DG, Feng FY, Iyer HK, Chinnaiyan AM. 2015.The landscape of long noncoding RNAs in the human transcriptome.Nature Genetics, 47(3): 199-208.
Kent WJ, Baertsch R, Hinrichs A, Miller W, Haussler D. 2003. Evolution′s cauldron: duplication, deletion, and rearrangement in the mouse and human genomes.Proceedings of the National Academy of Sciences of the United States of America, 100(20): 11484-11489.
Kim EB, Fang X, Fushan AA, Huang Z, Lobanov AV, Han L, Marino SM,Sun X, Turanov AA, Yang P, Yim SH, Zhao X, Kasaikina MV, Stoletzki N,Peng C, Polak P, Xiong Z, Kiezun A, Zhu Y, Chen Y, Kryukov GV, Zhang Q,Peshkin L, Yang L, Bronson RT, Buffenstein R, Wang B, Han C, Li Q, Chen L, Zhao W, Sunyaev SR, Park TJ, Zhang G, Wang J, Gladyshev VN. 2011.Genome sequencing reveals insights into physiology and longevity of the naked mole rat.Nature, 479(7372): 223-227.
Li R, Fan W, Tian G, Zhu H, He L, Cai J, Huang Q, Cai Q, Li B, Bai Y,Zhang Z, Zhang Y, Wang W, Li J, Wei F, Li H, Jian M, Li J, Zhang Z,Nielsen R, Li D, Gu W, Yang Z, Xuan Z, Ryder OA, Leung FC, Zhou Y, Cao J, Sun X, Fu Y, Fang X, Guo X, Wang B, Hou R, Shen F, Mu B, Ni P, Lin R,Qian W, Wang G, Yu C, Nie W, Wang J, Wu Z, Liang H, Min J, Wu Q,Cheng S, Ruan J, Wang M, Shi Z, Wen M, Liu B, Ren X, Zheng H, Dong D,Cook K, Shan G, Zhang H, Kosiol C, Xie X, Lu Z, Zheng H, Li Y, Steiner CC,Lam TT, Lin S, Zhang Q, Li G, Tian J, Gong T, Liu H, Zhang D, Fang L, Ye C, Zhang J, Hu W, Xu A, Ren Y, Zhang G, Bruford MW, Li Q, Ma L, Guo Y,An N, Hu Y, Zheng Y, Shi Y, Li Z, Liu Q, Chen Y, Zhao J, Qu N, Zhao S,Tian F, Wang X, Wang H, Xu L, Liu X, Vinar T, Wang Y, Lam TW, Yiu SM,Liu S, Zhang H, Li D, Huang Y, Wang X, Yang G, Jiang Z, Wang J, Qin N, Li L, Li J, Bolund L, Kristiansen K, Wong GK, Olson M, Zhang X, Li S, Yang H,Wang J, Wang J. 2010. The sequence and de novo assembly of the giant panda genome.Nature, 463(7279): 311-317.
Lindblad-Toh K, Garber M, Zuk O, Lin MF, Parker BJ, Washietl S,Kheradpour P, Ernst J, Jordan G, Mauceli E, Ward LD, Lowe CB, Holloway AK, Clamp M, Gnerre S, Alfoldi J, Beal K, Chang J, Clawson H, Cuff J, Di Palma F, Fitzgerald S, Flicek P, Guttman M, Hubisz MJ, Jaffe DB, Jungreis I, Kent WJ, Kostka D, Lara M, Martins AL, Massingham T, Moltke I, Raney BJ, Rasmussen MD, Robinson J, Stark A, Vilella AJ, Wen J, Xie X, Zody MC, Baldwin J, Bloom T, Chin CW, Heiman D, Nicol R, Nusbaum C, Young S, Wilkinson J, Worley KC, Kovar CL, Muzny DM, Gibbs RA, Cree A, Dihn HH, Fowler G, Jhangiani S, Joshi V, Lee S, Lewis LR, Nazareth LV,Okwuonu G, Santibanez J, Warren WC, Mardis ER, Weinstock GM, Wilson RK, Delehaunty K, Dooling D, Fronik C, Fulton L, Fulton B, Graves T, Minx P, Sodergren E, Birney E, Margulies EH, Herrero J, Green ED, Haussler D,Siepel A, Goldman N, Pollard KS, Pedersen JS, Lander ES, Kellis M. 2011.A high-resolution map of human evolutionary constraint using 29 mammals.Nature, 478(7370): 476-482.
Marinotti O, Cerqueira GC, De Almeida LGP, Ferro MIT, Loreto ELDS, Zaha A, Teixeira SMR, Wespiser AR, Almeida E Silva A, Schlindwein AD,Pacheco ACL, Silva ALDCD, Graveley BR, Walenz BP, Lima BDA, Ribeiro CaG, Nunes-Silva CG, De Carvalho CR, Soares CMDA, De Menezes CBA,Matiolli C, Caffrey D, Araújo DA, De Oliveira DM, Golenbock D, Grisard EC,Fantinatti-Garboggini F, De Carvalho FM, Barcellos FG, Prosdocimi F, May G, Azevedo Junior GMD, Guimar?es GM, Goldman GH, Padilha IQM,Batista JDS, Ferro JA, Ribeiro JMC, Fietto JLR, Dabbas KM, Cerdeira L,Agnez-Lima LF, Brocchi M, De Carvalho MO, Teixeira MDM, Diniz Maia MDM, Goldman MHS, Cruz Schneider MP, Felipe MSS, Hungria M, Nicolás MF, Pereira M, Montes MA, Cant?o ME, Vincentz M, Rafael MS,Silverman N, Stoco PH, Souza RC, Vicentini R, Gazzinelli RT, Neves RDO,Silva R, Astolfi-Filho S, Maciel TE, Urményi TP,, Tadei WP, Camargo EP,De Vasconcelos ATR. 2013. The Genome of Anopheles darlingi, the main neotropical malaria vector.Nucleic Acids Research, 41(15): 7387-7400.
Nene V, Wortman JR, Lawson D, Haas B, Kodira C, Tu ZJ, Loftus B, Xi Z,Megy K, Grabherr M, Ren Q, Zdobnov EM, Lobo NF, Campbell KS, Brown SE, Bonaldo MF, Zhu J, Sinkins SP, Hogenkamp DG, Amedeo P,Arensburger P, Atkinson PW, Bidwell S, Biedler J, Birney E, Bruggner RV,Costas J, Coy MR, Crabtree J, Crawford M, Debruyn B, Decaprio D,Eiglmeier K, Eisenstadt E, El-Dorry H, Gelbart WM, Gomes SL, Hammond M, Hannick LI, Hogan JR, Holmes MH, Jaffe D, Johnston JS, Kennedy RC,Koo H, Kravitz S, Kriventseva EV, Kulp D, Labutti K, Lee E, Li S, Lovin DD,Mao C, Mauceli E, Menck CF, Miller JR, Montgomery P, Mori A,Nascimento AL, Naveira HF, Nusbaum C, O′leary S, Orvis J, Pertea M,Quesneville H, Reidenbach KR, Rogers YH, Roth CW, Schneider JR,Schatz M, Shumway M, Stanke M, Stinson EO, Tubio JM, Vanzee JP,Verjovski-Almeida S, Werner D, White O, Wyder S, Zeng Q, Zhao Q, Zhao Y, Hill CA, Raikhel AS, Soares MB, Knudson DL, Lee NH, Galagan J,Salzberg SL, Paulsen IT, Dimopoulos G, Collins FH, Birren B, Fraser-Liggett CM, Severson DW. 2007. Genome sequence of Aedes aegypti, a major arbovirus vector.Science, 316(5832): 1718-1723.
Sanil D, Shetty V, Shetty NJ. 2014. Differential expression of glutathione stransferase enzyme in different life stages of various insecticide-resistant strains of Anopheles stephensi: a malaria vector.Journal of Vector Borne Diseases, 51(2): 97-105.
Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K,Clawson H, Spieth J, Hillier LW, Richards S, Weinstock GM, Wilson RK,Gibbs RA, Kent WJ, Miller W, Haussler D. 2005. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes.Genome Research, 15(8): 1034-1050.
Takken W & Verhulst NO. 2013. Host preferences of blood-feeding mosquitoes.Annual Review of Entomology, 58: 433-453.
Tian J, Zhao Z-H, Chen H-P. 2009. Conserved non-coding elements in human genome.Hereditas(Beijing), 31(11): 1067-1076.
Tsuji N, Okazawa T, Yamamura N. 1990a. Autogenous and anautogenous mosquitoes: a mathematical analysis of reproductive strategies.Journal of Medical Entomology, 27(4): 446-453.
Tsuji N, Okazawa T, Yamamura N. 1990b. Autogenous and anautogenous mosquitoes: a mathematical analysis of reproductive strategies.Journal of Medical Entomology, 27(4): 446-453.
Wang K, Li M, Hakonarson H. 2010. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data.Nucleic Acids Research, 38(16): e164.
Woolfe A, Goodson M, Goode DK, Snell P, Mcewen GK, Vavouri T, Smith SF, North P, Callaway H, Kelly K, Walter K, Abnizova I, Gilks W, Edwards YJ, Cooke JE, Elgar G. 2005. Highly conserved non-coding sequences are associated with vertebrate development.PLoS Biology, 3(1): e7.
You M, Yue Z, He W, Yang X, Yang G, Xie M, Zhan D, Baxter SW, Vasseur L, Gurr GM, Douglas CJ, Bai J, Wang P, Cui K, Huang S, Li X, Zhou Q, Wu Z, Chen Q, Liu C, Wang B, Xu X, Lu C, Hu M, Davey JW, Smith SM, Chen M, Xia X, Tang W, Ke F, Zheng D, Hu Y, Song F, You Y, Ma X, Peng L,Zheng Y, Liang Y, Chen Y, Yu L, Zhang Y, Liu Y, Li G, Fang L, Li J, Zhou X,Luo Y, Gou C, Wang J, Yang H. 2013. A heterozygous moth genome provides insights into herbivory and detoxification.Nature Genetics, 45(2):220-225.
Zeng YN, Shen YY, Zhang YP. 2013. Genome-wide scan reveals the molecular mechanisms of functional differentiation of Myotis lucifugus and Pteropus vampyrus.Zoological Research, 34(3): 221-227.
Zhang Y, Zhang XO, Chen T, Xiang JF, Yin QF, Xing YH, Zhu S, Yang L,Chen LL. 2013. Circular intronic long noncoding RNAs.Molecular Cell,51(6): 792-806.
Zhao L, Becnel JJ, Clark GG, Linthicum KJ. 2010. Expression of AeaHsp26 and AeaHsp83 in Aedes aegypti (Diptera: Culicidae) larvae and pupae in response to heat shock stress.Journal of MedicalEntomology, 47(3): 367-375.

- Zoological Research的其它文章
- Morphometric studies of genus Placocheilus (Teleostei:Cypriniformes) from Red River, China
- Patterns of reptile and amphibian species richness along elevational gradients in Mt. Kenya
- Stress-relevant social behaviors of middle-class male cynomolgus monkeys (Macaca fascicularis)
- Morphometric variability of Arctodiaptomus salinus(Copepoda) in the Mediterranean-Black Sea region
- Physiological approaches to understanding molecular actions on dorsolateral prefrontal cortical neurons underlying higher cognitive processing
- Editor’s comments