
Using NSGA-III for optimising biomedical ontology alignment
2019-10-18 03:10:06XingsiXueJiaweiLuJunfengChen
Xingsi Xue ?,Jiawei Lu ,Junfeng Chen
1College of Information Science and Engineering,Fujian University of Technology,Fuzhou,Fujian,People’s Republic of China
2Intelligent Information Processing Research Center,Fujian University of Technology,Fuzhou,Fujian,People’s Republic of China
3Fujian Provincial Key Laboratory of Big Data Mining and Applications,Fujian University of Technology,Fuzhou,Fujian,People’s Republic of China
4Fujian Key Laboratory for Automotive Electronics and Electric Drive,Fujian University of Technology,Fuzhou,Fujian,People’s Republic of China
5College of IOT Engineering,Hohai University,Changzhou,Jiangsu,People’s Republic of China
?E-mail:jack8375@gmail.com
Abstract:To support semantic inter-operability between the biomedical information systems,it is necessary to determine the correspondences between the heterogeneous biomedical concepts,which is commonly known as biomedical ontology matching.Biomedical concepts are usually complex and ambiguous,which makes matching biomedical ontologies a challenge.Since none of the similarity measures can distinguish the heterogeneous biomedical concepts in any context independently,usually several similarity measures are applied together to determine the biomedical concepts mappings.However,the ignorance of the effects brought about by different biomedical concept mapping’s preference on the similarity measures significantly reduces the alignment’s quality.In this study,a non-dominated sorting genetic algorithm (NSGA)-III-based biomedical ontology matching technique is proposed to effectively match the biomedical ontologies,which first utilises an ontology partitioning technique to transform the large-scale biomedical ontology matching problem into several ontology segment-matching problems,and then uses NSGA-III to determine the optimal alignment without tuning the aggregating weights.The experiment is conducted on the anatomy track and large biomedic ontologies track which are provided by the Ontology Alignment Evaluation Initiative (OAEI),and the comparisons with OAEI’s participants show the effectiveness of the authors’ approach.
1 Introduction
Over the recent years,ontologies have been extensively used in biomedical domains [1]such as annotation of medical records [2],medical knowledge representation and sharing [3],clinical data integration and medical decision making [4].The vast usage of ontologies in biomedical domain has compelled researchers to develop more biomedical ontologies such as gene ontology (GO)[5],National Cancer Institute (NCI) thesaurus [6],Foundation Model of Anatomy (FMA) [7]and the Systemised Nomenclature of Medicine (SNOMED-CT) [8].However,because of human subjectivity,various biomedical ontologies may use different terms for the same meaning or may use the same term to mean different things,yielding ontology heterogeneous problem.For example,when describing the muscles surrounding the human heart,NCI ontology uses the term ‘Myocardium’ but FMA utilises ‘Cardiac Muscle Tissue’.Thus,to integrate the knowledge regarding human heart,it is necessary for a biomedical system to determine the correspondences between NCI and FMA.Similarly,finding correspondence between GO and FMA can be used by molecular biologist in understanding the outcome of proteomics and genomics in a large-scale anatomic view [9].Moreover,the correspondences between ontologies have also been used for heterogeneity resolution among various health standards [10].The biomedical concept mapping set between two ontologies is called the alignment and the process of discovering it is termed as ontology matching.
Matching biomedical ontologies is an open challenge in the ontology matching domain because biomedical concepts are usually complex and ambiguous.Frequently,the same entity has several names (e.g.gluconeogenesis,glucose synthesis and glucose biosynthesis,all refer to the same metabolic process),a common word refers to a biomedical concept (e.g.hedgehog and fruity are both gene names) or even the same word can be applied to two different biomedical concepts (e.g.lingula can either be a structure of the brain or the lung).Since none of the similarity measures can distinguish the same biomedical concepts in any contexts independently,the ontology matching systems actually apply several similarity measures to determine the correspondences between particular biomedical concepts.The most common composition of multiple similarity measures is the parallel composition,where the similarity measures are executed independently from each other and the aggregated correspondence is computed afterwards [11].Currently,researchers mainly focus on how to tune the aggregating weights for various similarity measures to improve the quality of the ontology alignments [12].However,the ignorance of the effects brought about by different biomedical concept mapping’s preferences on some similarity measures significantly reduce the alignment’s quality.For example,it is better to use the linguistic-based similarity measure instead of syntactic-based similarity measure to distinguish two terms ‘Myocardium’ and ‘Cardiac Muscle Tissue’,and weights tuned in this way could be problem specific,which means they might not be reused in other matching scenarios.Moreover,existing matching techniques can only deal with small-scale ontologies,and their runtime and memory consumption are always long and huge when matching biomedical ontologies which often possess tens of thousands of concepts.To effectively match the biomedical ontologies,in this paper,we propose a non-dominated sorting genetic algorithm (NSGA)-III-based [13]ontology matching technique to optimise the biomedical ontology alignment.In particular,the contributions made in this paper are as follows:
? A large-scale biomedical ontology matching framework is proposed.
? A many-objective optimal model is constructed for the biomedical ontology matching problem.
? A problem-specific NSGA-III has presented to optimise the biomedical ontology alignment,which can improve the convergence as well as maintain the diversity during the matching process.
The rest of this paper is organised as follows:Section 2 describes the related works;Section 4 shows the biomedical ontology partitioning technique;Section 5 defines many-objective similarity measure combining problem and presents the NSGA-III-based ontology matching technique;Section 6 presents the experimental studies and analysis;and finally,Section 7 draws the conclusions and presents the future work.
2 Related work
In general,the basic similarity measures can be divided into three broad categories,i.e.syntactic-based similarity measure,linguistic-based similarity measure and structure-based similarity measure.In particular,syntactic-based similarity measure computes the edit distance between ontology entities such as similarity measure for ontology alignment (SMOA) [14].Linguistic-based matcher utilises synonymy,hypernymy and other linguistic relations to calculate the similarity score between ontology entities which require a lexicon and thesauri such as WordNet [15].Structure-based matcher computes a similarity score between two ontological entities based on their ontology taxonomy hierarchy structure,and the common intuition is that two distinct ontology entities are similar when their adjacent entities are similar.The most popular structure-based similarity measures are the well known similarity flooding (SF) algorithm[16]and the profile-based similarity measure [12].Although both of them utilise the ontology’s taxonomy structure to calculate the similarity value,SF executes an iterative fix-point computing process,while the profile-based similarity measure first constructs for each entity a profile by collecting the data properties from its direct descendants and itself,then,the similarity value between two entities is measured by calculating the similarity of their corresponding profiles.
Usually,similarity measure combination and tuning are tackled by setting appropriate weight set through different methods.The most outstanding approach in this area is COMA++[17]which utilises two kinds of similarity measures:simple similarity measure such as the syntactic-based similarity measure and linguistic-based similarity measure and hybrid similarity measure that combines multiple similarity measures.COMA++’s aggregating weights are determined by an expert.Lately,the focus is placed on the heuristic techniques for combining different similarity measures.The first method is called harmonic adaptive weighted sum which is presented in the PRIOR+[18].The harmony value is calculated through a similarity matrix and further assigned as the weight to the similarity measure associated with that matrix.PRIOR+integrates the syntactic-based similarity measure and structurebased similarity measure.The second method is called the local confidence weighted sum,which is the core method for combining individual similarity measures in the AgreementMaker [19].This measure is defined for an entity by considering the average of similarity values of entities that are associated (or not associated)with that entity.Finally,the selection of the final candidates from the set of candidates is performed by a greedy selection strategy.In particular,AgreementMaker utilises the syntactic-based similarity measure and linguistic-based similarity measure.For a given matching scenario,YAM++[20]evaluates the degree of reliability of these similarity measures and assigns appropriate weight values to them.More recently,Benaissa and Khiat [21]propose a heuristic strategy to estimate the weights for different similarity measures,which is of a statistical nature and estimates the weights by an estimation of the precision standard metric.Particularly,the similarity measures they use are the linguisticbased similarity measure and structure-based similarity measure.
Recently,evolutionary algorithms (EAs) are appearing as an effective methodology to determine the optimal aggregating weights for different similarity measures.Genetic algorithm based ontology alignment (GOAL) [22]is the first matching system that utilises EA to determine the optimal weight configuration for a weighted average aggregation of several similarity measures by considering a reference alignment.A similar idea of combining multiple similarity measures is also developed by Naya et al.[23],Alexandru Lucian and Iftene[24]and Guli′c et al.[11].To improve efficiency,a hybrid EA is presented to tune the parameters for aggregating various similarity measures [12,25].More recently,Xue and Liu [26]present an approach based on a multi-objective EA to determine the optimal weights being assigned to the profile-based similarity measure,WordNet-based similarity measure and structure-based similarity measure.All these methods dedicate to tune the weights for aggregating different similarity measures,which ignore the effects brought about by different entity mappings’ preferences on different similarity measures,and thus,decrease the quality of the alignment.In this work,a many-objective matching technique is proposed to further improve the alignment’s quality,which takes into consideration each mapping’s preference on various similarity measures and determine the optimal alignment without tuning the aggregating weights.
3 Large-scale biomedical ontology matching framework
The proposed large-scale biomedical ontology matching framework is shown in Fig.1.As shown in this figure,our proposal first utilises an ontology partitioning technique to transform the biomedical ontology matching problem into several ontology segmentmatching problems and then uses NSGA-III to combine various similarity measures and optimise the quality of the ontology alignment.The former technique can transform the large-scale biomedical ontology matching problem into several ontology segment-matching problems,which can improve the efficiency of the matching process hereafter.The latter can trade-off each biomedical concept mapping’s preference on various similarity measures,and determine the optimal alignment without tuning the aggregating weights.Finally,the segment alignments are aggregated into a final alignment which is further evaluated with the reference alignment.
4 Biomedical ontology partition
Partitioning the large-scale biomedical ontology into various segments,where the term ‘segment’ is referred to as a fragment of an ontology,is an efficient way of reducing the algorithm’s search space [27].In this work,an alignment-oriented ontology partition technique [28]is introduced to partition the ontologies into various similar ontology segment pairs.First of all,the ontology with better reliability is selected as the source ontology.The reliability of an ontology is measured by the semantic accuracy,which is computed through the average of the squared semantic distance between each concept ciand the ontology O’s taxonomic root node ROOT.In particular,the formula for calculating semantic accuracy is presented as follows:

The source ontology is partitioned into disjoint segments through an ontology partition algorithm which is extended from structural clustering algorithm for network (SCAN) [29].Then,a concept relevance measure-based approach is adopted to determine the similar target ontology segments of each source ontology segment segsrc.Particularly,for each target ontology concept ci,the similarity value simcibetween ciand segsrcis calculated by summing up every SMOA(ci,cj) (see also Section 7.1).If simciis larger than the threshold,ciwill be added to the candidate concept set Ccand.If the relevance value of a concept in Ccandis bigger than the threshold,it will be added to the final target segment.Given a concept cmCcand,the relevance value of cmto source ontology segment can be calculated by the following formula:
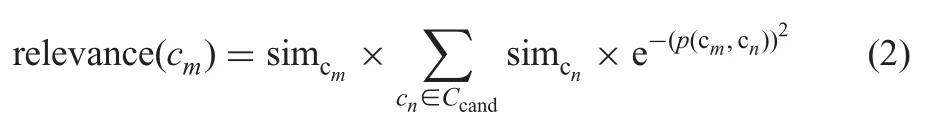
where simcmand simcn,respectively,denote the similarity values of cmand cnto segsrcand p(cm,cn)is the shortest length between their corresponding vertexes in ontology taxonomy structure.
After partitioning the ontologies,the matching process only needs to deal with the similar biomedical ontology segments’ matching problem,and all the similarity values obtained in the process of ontology partitioning are stored in the hash map to avoid repeating calculations in the hereafter matching process.With respect to the details of the alignment-oriented ontology partition algorithm,please see also [30].
5 Many-objective similarity measure combination
5.1 Many-objective similarity measure combining problem
Although the alignment evaluation measures recall,precision and f-measure [31]can reflect the quality of the resulting alignment,the reference alignment between two ontologies is usually unknown for real-life match problems [32].In this work,based on the observations that the more correspondences found and the higher mean similarity values of the correspondences are,the better the alignment quality is [33],we utilise the following metric to measure the quality of an alignment:
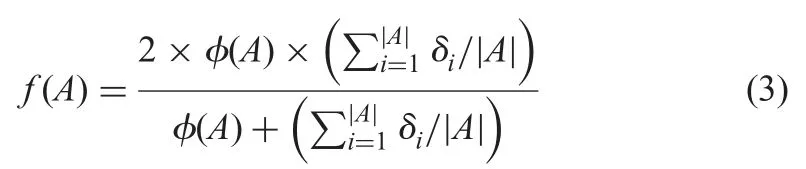
where|A|is the number of correspondences in A;fis a function of normalisation in [0,1];anddiis the similarity value of the ith correspondence in A.
On this basis,the many-objective optimal model of combining various similarity measures can be defined as follows:
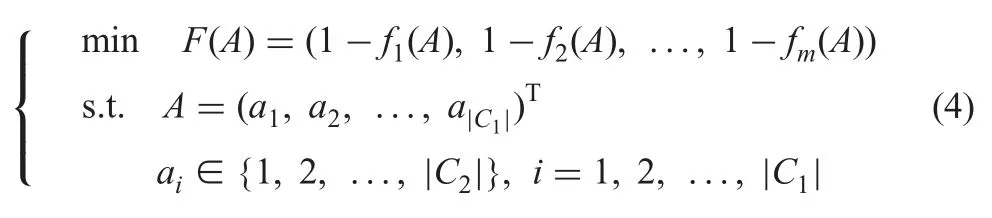
where m is the number of similarity measures;fi(A),i=1,2,...,m,calculates the alignment A’s quality with respect to the ith similarity measure;|C1| and |C2|,respectively,represent the cardinalities of source concept set C1and target concept set C2;and ai,i=1,2,...,|C1| represents the ith pair of correspondence.
Similarity measure takes as input two concept sets C1and C2and output a |C1|×|C2| similarity matrix S,whose element sijis the similarity score between the ith concept in |C1| and the jth concept in |C2|.Since the number of elements in biomedical ontology is large,we should avoid allocating an n1×n2similarity matrix,where n1and n2are the cardinalities of two concept sets.On the basis of the observation that a correct alignment should be consistent with the concept hierarchies organised by ‘is-a’ [34],if two concepts c1and c2have high similarity value,so-called anchors in the partitioning process,the sub-concepts(/super-concepts) of c1and super-concepts (/sub-concepts) of c2can be skipped or directly set as 0.Then,considering the similarity matrix is a typical sparse matrix,the compression techniques can be further adopted to replace it.It usually compresses a similarity matrix into several mega bytes (MBs).In our approach,we first replace the two-dimensional (2D) reduction set with 1D style,then merge the continuous number of elements as a link.
5.2 NSGA-III for optimising biomedical ontology alignment
NSGA-III is a many-objective algorithm proposed by Deb et al.[35],which introduces a well-distributed reference points based clustering operator to replace the crowding distance operator in NSGA-II.In this work,NSGA-III [13]is utilised to automatically combine various similarity measures and determine the optimal biomedical ontology segment alignment.Original NSGA-III emphasises that the solutions should be Pareto non-dominated and closed to the reference line of each reference point.However,with the growing number of the objectives,selection pressure based on Pareto dominance would be too small to pull the population toward Pareto front,and in this case,NSGA-III indeed emphasises diversity more than convergence.To this end,we present a problem-specific NSGA-III to improve the convergence as well as maintain the diversity when matching the biomedical ontology segments.
Next,three key components of NSGA-III are presented in details,i.e.encoding mechanism,uniform design-based reference points generation andu-dominance.Finally,the outline of problemspecific NSGA-III is given.
5.2.1 Encoding mechanism:Let |C1| and |C2| be the cardinalities of the source concept set C1and target concept set C2,respectively.Each chromosome in the population would be a 1D array with |C1| elements,and the elements are denoted as:N1N2···N|C1|,where Ni{0,1,...,|C2|},i{1,...,|C1|},which means the ith concept in C1is mapped to the Nith concept in C2.In particular,when Ni=0,the ith concept is not mapped to any concept in C2.
5.2.2 Uniformly distributed reference points:In the original NSGA-III,the Das and Dennis’s systematic approach[36]is used to generate reference points.However,when the number of objectives is high,the number of reference points generated by this approach would become very large [37].In our work,we propose to use a uniform design [38],which aims at determining a set of points that are uniformly distributed over the design space,to produce uniformly distributed reference points in a unit sphere S=First,we need to generate a set of Q uniformly distributed points on C={(c1,c2,...,cm)|0 ≤c1,c2,...,cm≤1}.Let Q be the number of uniform distributed points in C;m be the dimension of the problem that is equal to the number of basic similarity measures in this work;dbe the number that yields the smallest discrepancy of generated point set (see also [39]),an integer matrix so-called uniform array [Mij]Q×mcan be calculated with Mij=idj?1mod Q+1,i=1,2,...,Q,j=1,2,...,m,where the ith row of it can define a point Ci=(ci,1,ci,2,...,ci,m) with cij=(2Mij?1)/2Q,i=1,2,...,Q,j=1,2,...,m.Next,a set of Q reference points uniformly distributed on S,denoted by P(Q,m)=Pi=(pi,1,pi,2,...,pi,m),can be calculated as follows:
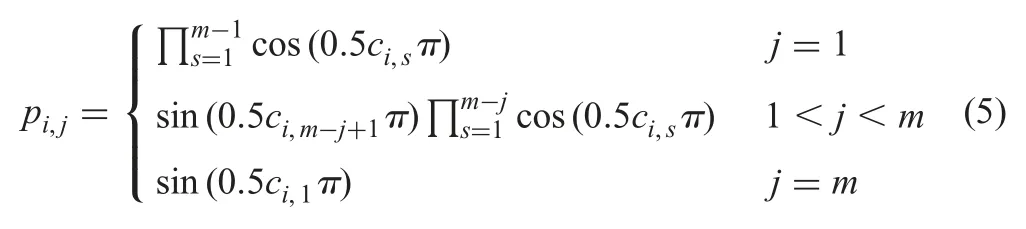
Equation(5)is a hyper-sphere formula,and in particular,it becomes a circular formula when m=2 and a spherical formula when m=3.
5.2.3u-Dominance:Given reference points P(Q,m) which can be denoted by {Pi,P2,...,PQ},a reference line is defined by joining a reference point with the origin.After that,each individual is associated with a reference point by calculating the perpendicular distance of it from each of the reference line.The reference point whose reference line is closest to a solution is considered to be associated with this solution.In this way,the population can be split into Q clusters C={C1,C2,...,cQ}where the cluster Cjis represented by the reference point Pj,j=1,2,...,Q.
Given a solution x and its objective vector f(x) which can be denoted by [f1(x),f2(x),...,fm(x)],reference line Ljpassing through the origin point Z and Pi,a penalty function [40]can be defined aswherecalculates the perpendicular distance between f(x) and Lj
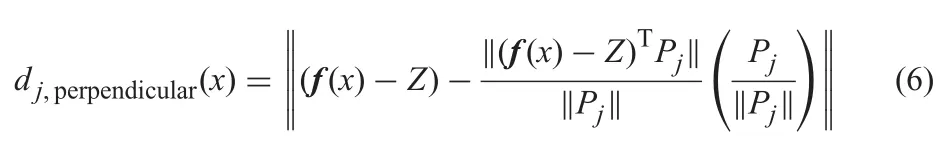
Given m=2,an example of the perpendicular distance is shown in Fig.2.
In this work,u.0 is a predefined penalty parameter,which is set as 2 to achieve the best mean quality of alignment on all testing cases.It is obvious that the smallerf(x)and dj,perpendicular(x),respectively,lead to better convergence and better diversity.Given two solutions x,yV,x is said tou-dominate y,denoted by x ?uy,if x,yCjand Dj(x),Dj(y),j{1,2,...,Q} [37].Then,we utilise theu-dominance to implement the fast non-dominated sorting [35]on the population to partition it into differentu-non-domination levels.
5.2.4 Flowchart of NSGA-III:The flowchart of NSGA-III is presented in Fig.3.First,we apply a uniform design-based method to generate any number of reference points,and the common one point crossover operator and the bit mutation operator.Before calculating the perpendicular distance between a population and each of the reference lines,NSGA-III needs to normalise objectives’ values and supplied reference points,which can ensure they have an identical range.In this work,since all the objective’s values are in the same range [0,1]and the ideal point is the zero vector,we do not need to carry out the normalisation in each generation.In addition,replace the Pareto dominance in NSGA-III withu-dominance to trade-off the convergence and diversity in many-objective optimisation,and utilise theu-dominance based fast non-dominated sorting is employed on the population clusters to divide them into differentu-non-domination levels.Finally,we determine the next generation’s population by including oneu-non-domination at a time,which starts from the first level.With respect to the solutions in the last accepted level,we first sort them in ascending order according to their mean f() values and then select the solutions sequentially.In this work,in order to compare with other ontology matching systems whose results are measured with f-measure,we pick up the solution in the Pareto front with the highestas the representative solution.
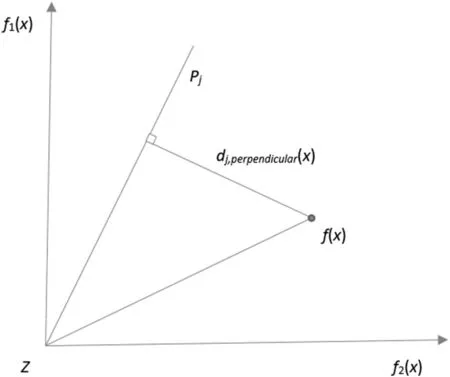
Fig.2 Example of the perpendicular distance
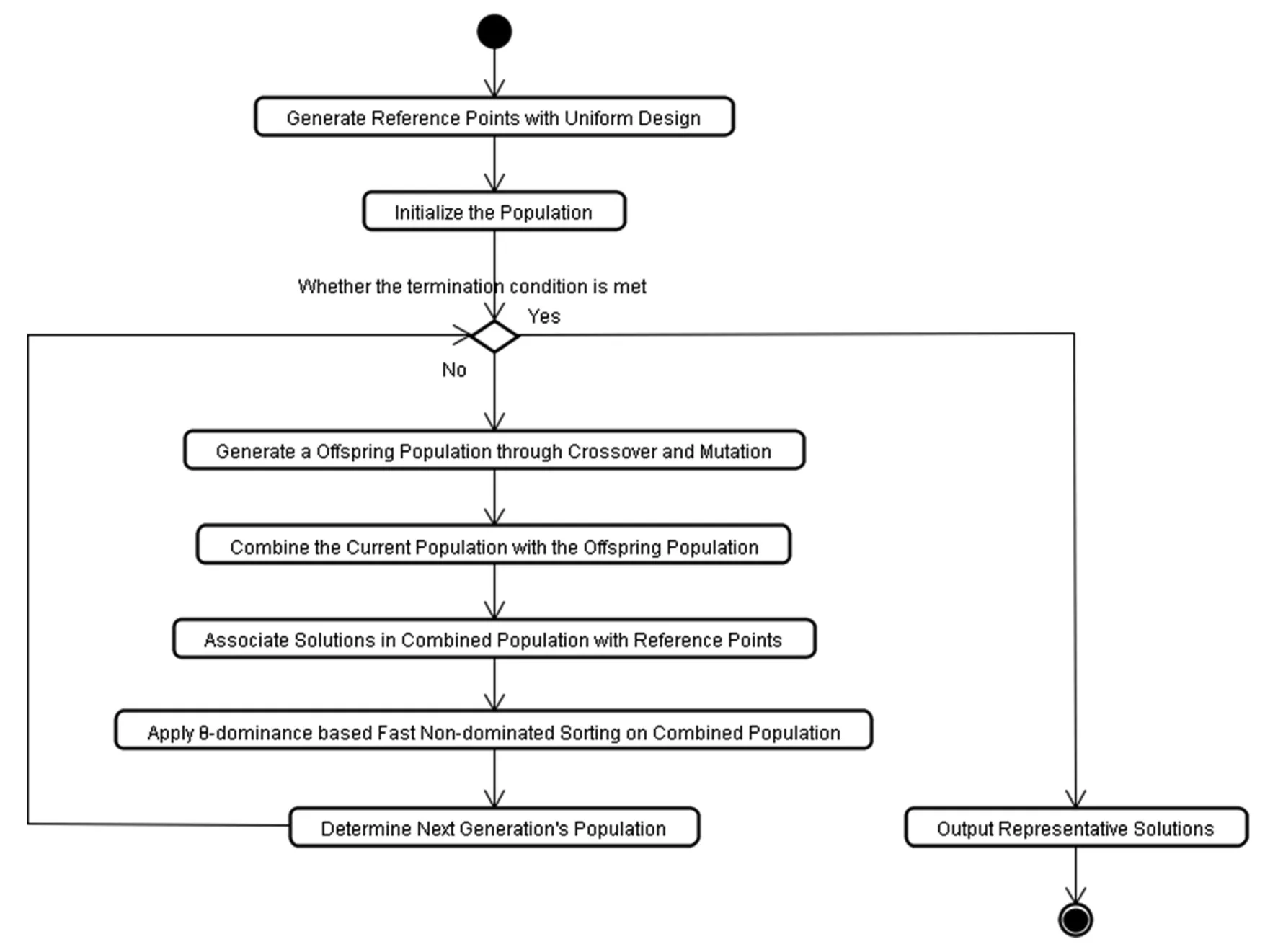
Fig.3 Flowchart of NSGA-III
6 Experimental studies and analysis
In this work,we exploit the Anatomy[http://oaei.ontologymatching.org/2017/anatomy/index.html.]and Large Biomed [http://www.cs.ox.ac.uk/isg/projects/SEALS/oaei/2017/.]track to study the effectiveness of our approach,which are provided by Ontology Alignment Evaluation Initiative (OAEI) 2017 [http://oaei.ontologymatching.org/2017.].Tables 1 and 2 show the mean value of f-measure of the alignments obtained by our approach in 30 independent runs and the results obtained by the participants of OAEI.
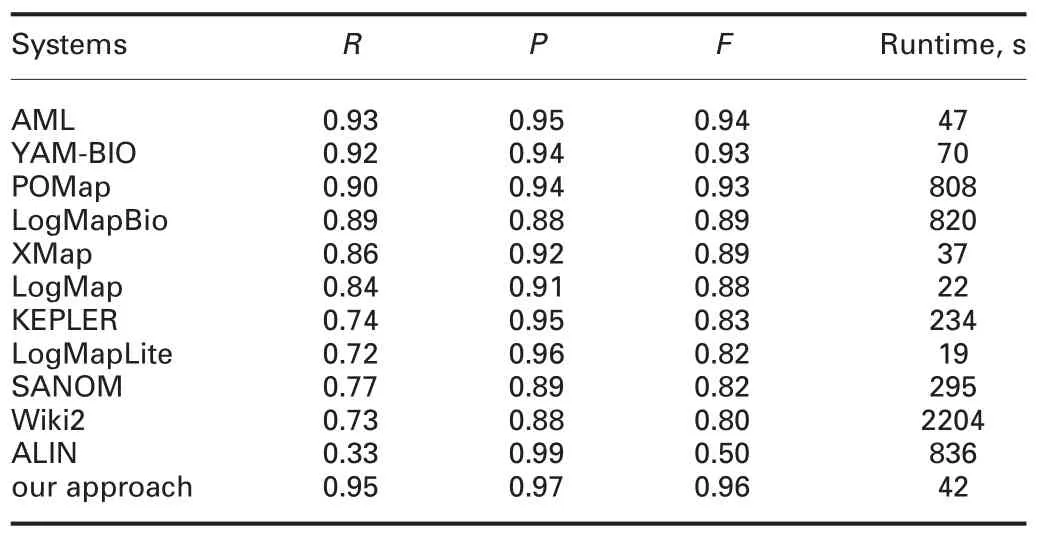
Table 1 Comparison of anatomy track in OAEI 2017
Three main categories of similarity measures are utilised in this work,i.e.SMOA (a syntactic-based similarity measure),Unified Medical Language System-based [41]similarity measure(a linguistic-based similarity measure) and profile-based similarity measure (a structure-based similarity measure) [12].The parameters used by NSGA-III are as follows:numerical accuracy=0.01,number of reference points=20,population size=25,crossover probability=0.8,mutation probability=0.02and maximum number of generation=300.These parameters represent a trade-off setting obtained in an empirical way to achieve the highest average alignment quality on all test cases of the exploited dataset,which is robust against the heterogeneous situations in our experiment.
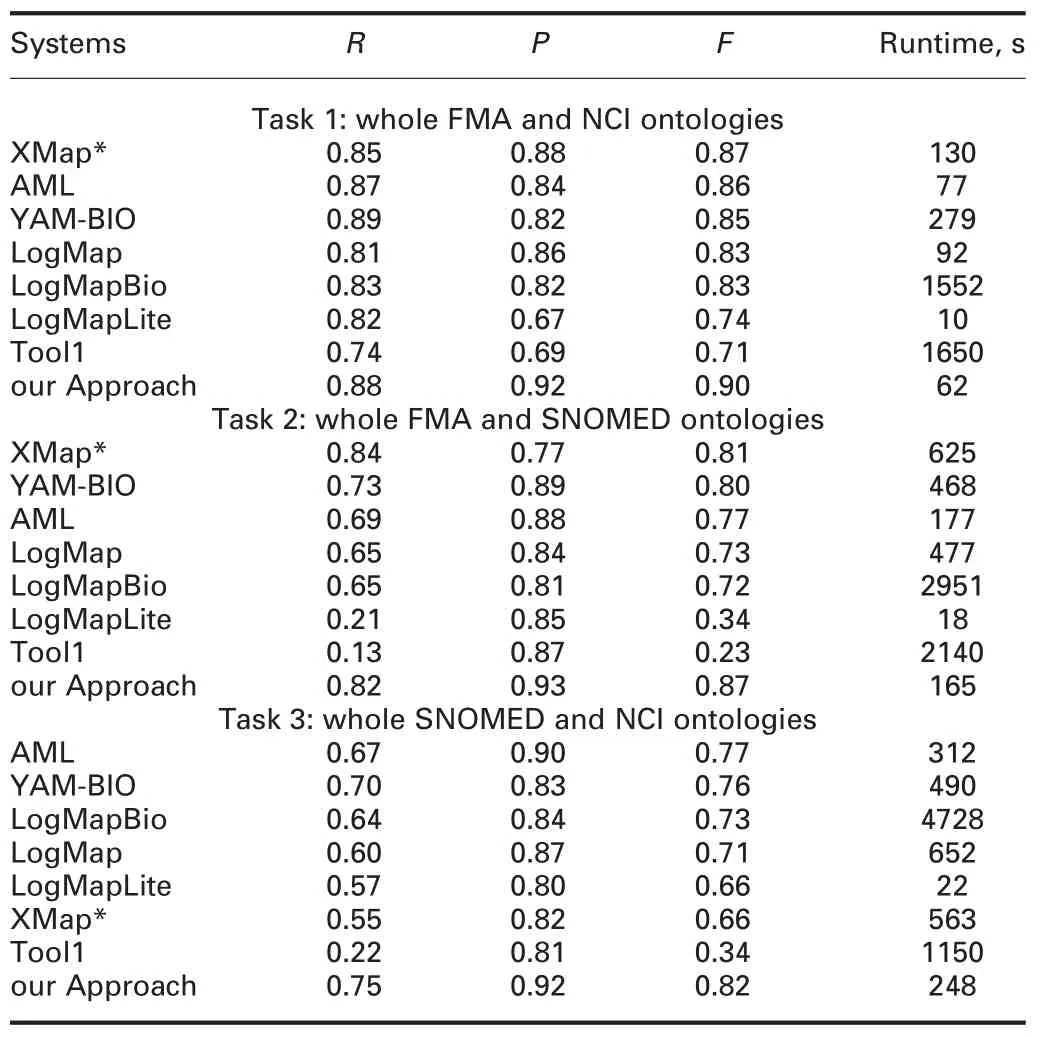
Table 2 Comparison of large Biomed track in OAEI 2017
We run the anatomy track with a CPU @ 3.46 GHz×6 with 8 GB allocated RAM,and the large biomed track with an Intel Core i9-8950HK CPU @ 2.90 GHz×12 and 25 GB allocated RAM,which is the same with the OAEI’s hardware configurations.
6.1 Anatomy track
The anatomy track is a large ontology matching task which is about matching the Adult Mouse Anatomy(2744 classes)and a part of the NCI thesaurus(3304 classes)describing the human anatomy.As can be seen from Table 1,our approach’s f-measure is the best among all the participants in OAEI 2017,and the runtime taken by our approach is 42 s,which is less than AgreementMakerLight(AML),the best matcher of OAEI 2017 on Anatomy track.In this track,our approach’s recall and precision are,in general,high,which further indicates the effectiveness of our approach.
6.2 Large biomedic ontologies track
This track aims at finding alignments between the large and semantically rich biomedical ontologies FMA,SNOMED-CT and NCI,which contains 78,989,306,591 and 66,724 classes,respectively.The track has been split into three matching problems:FMA–NCI,FMA–SNOMED and SNOMED–NCI,and each matching problem in three tasks involving different fragments of the input ontologies.
As can be seen from Table 1,in terms of f-measure and running time,our approach’s results are the best in all three tasks.In this track,our approach outperforms AML,which is the top ontology matcher and developed primarily for the biomedical ontology matching,in all three tasks in terms of f-measure,and the runtime of our approach is also less than AML.The experimental results further show the effectiveness of our proposal when matching large-scale biomedical ontologies.
7 Conclusion and future work
An ontology matching framework is proposed to efficiently match biomedical ontologies,which first uses an ontology partition technique to reduce the matching algorithm’s search space,and then utilises an NSGA-III-based biomedical ontology matching technique to directly determine the optimal alignment without tuning the aggregating weights.The experimental results show that our proposal is able to efficiently determine the high-quality biomedical ontology alignments.In continuation of our research,we are interested in combining more similarity measures.Moreover,some strategies which could remove the mappings that lead to logical conflicts can be introduced to further improve the alignment’s quality.
In the future,we are interested in getting the user involved in our approach to guide the search direction,so that the alignment quality could be further improved.Since the similarity measures would lead to the opposing results on the same biomedical concepts,before combining them,we need to select the effective similarity measures based on the heterogeneous characteristics of biomedical ontologies.How to select,combine and tune these similarity measures to improve the alignment’s quality is a challenge especially when the scale of similarity measures is huge.Therefore,we are also interested in carrying out a future study on such situation as combining more than 50 similarity measures to improve our proposal.
8 Acknowledgments
This work was supported by the National Natural Science Foundation of China (Nos.61503082 and 61403121),the Natural Science Foundation of Fujian Province (No.2016J05145),the Fundamental Research Funds for the Central Universities(No.2015B20214),the Program for New Century Excellent Talents in Fujian Province University (No.GY-Z18155),the Program for Outstanding Young Scientific Researcher in Fujian Province University (No.GY-Z160149) and the Scientific Research Foundation of Fujian University of Technology(No.GY-Z17162).
 CAAI Transactions on Intelligence Technology2019年3期
CAAI Transactions on Intelligence Technology2019年3期
- CAAI Transactions on Intelligence Technology的其它文章
- Imputing missing values using cumulative linear regression
- Multi-objective linear fractional inventory model with possibility and necessity constraints under generalised intuitionistic fuzzy set environment
- Advances on QoS-aware web service selection and composition with nature-inspired computing
- TLBO with variable weights applied to shop scheduling problems
- Expanded models of the project portfolio selection problem with learning effect
- Study on covering rough sets with topological methods