
Immunosuppressant treatment for IgG4-related sclerosing cholangitis: A case report
2021-02-22 06:20:26JongSunKimWonHoChoiKyungAnnLeeHyunSookKim
World Journal of Clinical Cases 2021年1期
Jong-Sun Kim, Won Ho Choi, Kyung-Ann Lee, Hyun-Sook Kim
Jong-Sun Kim, Won Ho Choi, Kyung-Ann Lee, Hyun-Sook Kim, Department of Internal Medicine,Soonchunhyang University Seoul Hospital, Seoul 04401, South Korea
Abstract BACKGROUND Immunoglobulin G4-related disease (IgG4-RD) is a multi-system fibroinflammatory disorder that can involve any organ, including the salivary glands,pancreas, and biliary tree. Treatment of immunoglobulin G4-related sclerosing cholangitis (IgG4-SC) is similar to that for IgG4-RD, but progression is irreversible in some cases. We present a case of IgG4-SC in which an immuno-suppressant induced marked clinical and radiologic improvement.CASE SUMMARY A 63-year-old male presented with a prominent itching sensation and wholebody jaundice. He showed obstructive-pattern jaundice, an elevated IgG4 level, and infiltration of a large number of IgG4-positive cells in the ampulla of Vater. The imaging findings of intrahepatic duct (IHD) and common bile duct dilation, an elevated serum IgG4 level, and characteristic histological findings led to diagnosis of IgG4-SC that compatible with the 2019 ACR/EULAR classification criteria. We planned to treat the patient with high-dose glucocorticoid (GC), followed by cyclophosphamide pulse therapy. After treatment with high-dose GC and an immunosuppressant, imaging studies showed that IHD dilatation had completely resolved.CONCLUSION Prompt diagnosis and appropriate treatment of IgG4-SC are important. Because there is a risk of relapse of IgG4-SC, the GC dose should be gradually reduced,and a maintenance immunosuppressant should be given.
Key Words: Immunoglobulin G4-related disease; Immunoglobulin G4-related sclerosing cholangitis; Glucocorticoid; Immunosuppressant; Case report; ACR/EULAR classification criteria
INTRODUCTION
Immunoglobulin G4-related disease (IgG4-RD) is an immune-mediated disorder that can induce fibrosis in multiple organs[1]. IgG4-related sclerosing cholangitis (IgG4-SC)is the most common extrapancreatic manifestation of IgG4-RD, and it is the third distinct entity of sclerosing cholangitis[2]. Treatment of IgG4-SC is similar to that for IgG4-RD involving autoimmune pancreatitis (AIP), but progression is irreversible in some cases. We demonstrate a case of IgG4-SC in which an immunosuppressant induced marked clinical and radiologic improvement.
CASE PRESENTATION
Chief complaints
A 63-year-old male was admitted to our hospital with an uncontrolled itching sensation and whole body jaundice.
History of present illness
Six months prior he had experienced intractable itching, which led to suspicion of obstructive cholangitis. Despite endoscopic retrograde cholangiopancreatography(ERCP) and endoscopic retrograde biliary drainage, which ruled out malignancy, the itching became aggravated and obstructive-pattern jaundice developed.
History of past illness
The patient’s medical history was non-specific with the exception of hypertension and diabetes mellitus, which were well controlled by medication. He was ex-smoker of ten years and a social drinker.
Personal and family history
No previous illnesses were reported and there was no family history of oncologic and rheumatic diseases.
Physical examination
The physical examination revealed icteric sclerae with multiple itching scratches on the skin.
Laboratory examinations
The patient’s laboratory results were as follows: Hemoglobin 10 g/dL (normal, 13-17 g/dL), hematocrit 30.6% (normal 39%-52%), platelet count 348000/L (normal, 130000-450000/L), aspartate aminotransferase 71 U/L (normal, 0-40 U/L), alanine aminotransferase level 44 U/L (normal, 0-41 U/L), gamma-glutamyl transferase level 233 U/L (normal, 5-61 U/L), alkaline phosphatase level 315 U/L (normal, 35-130 U/L), total bilirubin level 8.6 mg/dL (normal, 0-1.2 mg/dL), BUN level 26.7 mg/dL(normal, 6-20 mg/dL), creatinine level 1.37 mg/dL (normal, 0.5-1.2 mg/dL),erythrocyte sedimentation rate 120 mm/h (normal, 0-20 mm/h), C reactive protein(CRP) level 1.81 mg/dL (normal, 0-0.5 mg/dL), antinuclear antibody titer 1:2560(nucleolar pattern), IgG level 2416 mg/dL (normal, 700-1600 mg/dL), and serum IgG4 level 465 mg/dL (normal, 0-135 mg/dL).
Imaging examinations
Abdominal computed tomography (CT) showed intrahepatic duct (IHD) dilatation with intraductal soft tissue attenuation and periductal enhancement (Figure 1A). The common bile duct (CBD) was dilated with mild luminal narrowing but without findings of AIP (Figure 1B). There was no definite obstructive lesion or suspected malignancy. CT also showed fibrotic enhancement around the infrarenal aorta.
HISTOLOGICAL ASSESSMENT OF BIOPSY SPECIMEN
We performed ERCP to obtain an ampulla of Vater (AoV) biopsy for histologic analysis. The AoV biopsy revealed infiltration of inflammatory cells and granulation and many plasma cells in subepithelial stroma (Figure 2A and B). Immunohistochemical staining revealed a large number of infiltrating IgG4-positive cells(Figure 2C).
FINAL DIAGNOSIS
The imaging findings of IHD and CBD dilation, an elevated serum IgG4 level, and characteristic histological findings led to diagnosis of IgG4-SC.
TREATMENT
We planned to treat the patient with high-dose systemic glucocorticoid (GC,prednisolone 0.5 mg/kg/d), followed by cyclophosphamide pulse therapy (1000 mg/mo, 6 mo).
OUTCOME AND FOLLOW-UP
After 3 mo, CT showed that the IHD dilatation with intraductal soft tissue attenuation was almost completely resolved (Figure 1C). CT also showed slight regression of the CBD dilatation with mild luminal narrowing (Figure 1D) and no significant change in the soft tissue attenuation along the infrarenal abdominal aorta. Moreover, the total bilirubin and CRP levels had returned to within the normal ranges. On this basis we tapered the GC to 10 mg/d and added mycophenolate mofeltil (1000 mg/d) for maintenance (Figure 3). There was no recurrence of itching or jaundice at the 1 year follow up. Patient has provided informed consent for publication of the clinical process and images.
DISCUSSION
IgG4-RD has an immune-mediated fibrotic progression involving multiple organs and has myriad clinical manifestations[2,3]. IgG4-SC is a biliary manifestation of the systemic disorder IgG4-RD. IgG4-SC occurs in one-third of patients with type 1 AIP[4]. The prevalence of isolated IgG4-SC is less than 10% that of IgG4-RD[5]. The cholangiographic findings of IgG4-SC involving the hila and/or IHD are similar to those of hilar cholangiocarcinoma or primary sclerosing cholangitis (PSC). Therefore, it is important to differentiate IgG4-SC from the latter two conditions[6]. PSC is a chronic progressive disease with a poor prognosis and does not respond to GC therapy. In our case, IgG4-SC was not accompanied by AIP and could be differentiated from cholangiocarcinoma and PSC.
The Histology, Imaging, Serology, other Organ involvement and Response to therapy (HISORt) criteria of the Mayo Clinic are widely used for diagnosis of IgG4-RD, including IgG4-SC. The HISORt criteria include positive imaging findings,characteristic histological presentations, elevated serum IgG4 level, extrabiliary organ involvement, and a response to GC treatment[5]. The ACR/EULAR published new classification criteria for confident decision into more homogeneous group. The classification process comprises three steps. First, a potential IgG4 RD case must show involvement of at least 1 of 11 possible organs. Second, the case must not meet any of the 32 clinical, serologic, radiologic, and pathologic exclusion criteria. Third, the case must meet eight weighted inclusion criteria[7]. A threshold of 20 points has a specificity of 99.2% and a sensitivity of 85.5% for diagnosis of IgG4 RD; our patient’s score was 48 points. That score comprised the following: Histopathology, 6 points (lymphocytic infiltrate and obliterative phlebitis); immunostaining, 15 points (IgG4:IgG ratio 41%-70% and > 10 IgG4-positive cells/high power field); serum IgG4 concentration, 6 points (two- to five-fold the upper limit of normal); pancreas and biliary tree, 19 points(pancreas and biliary tree involvement); and retroperitoneal involvement, 4 points(diffuse thickening of the abdominal aortic wall). Thus, our patient met both the HISORt and the 2019 ACR/EULAR classification criteria.
The goal of IgG4-SC treatment is to induce rapid regression and to prevent irreversible fibrosis and biliary stricture. The majority of IgG4-RD patients respond to GC therapy. In this case the initial dose was prednisolone 30 to 40 mg/d for 1 mo,followed by tapering over 2 wk, then tapering of 5 mg/d for 3 to 4 mo. The final maintenance prednisolone dose was 5 mg/d. In some IgG4-SC case series, complete resolution of strictures and/or normalization of liver function were reported in up to 67% of the patients[8]. Patients with IgG4-SC are at high risk of relapse; the relapse rate is around 57%. Proximal strictures and a high serum IgG4 level are predictive of relapse. The maintenance option is typically low-dose corticosteroid, with or without immunosuppressive therapy with azathioprine or mycophenolate mofetil[9,10]. The treatment options for IgG4-SC relapse include increasing the dose of, or reintroducing,GC and addition of a second-line immunosuppressant; rituximab is an effective alternative and can lead to both substantial reduction of serum IgG4 concentration and clinical improvement. This agent works by depleting the pool of B cells that replenish the IgG4-secreting plasma cells[11]. In a prospective study of IgG4-RD, the rituximab response rate was 97%, and 77% and 46% of the patients were able to stop GC therapy and had not relapsed after 6 and 12 mo, respectively[12].
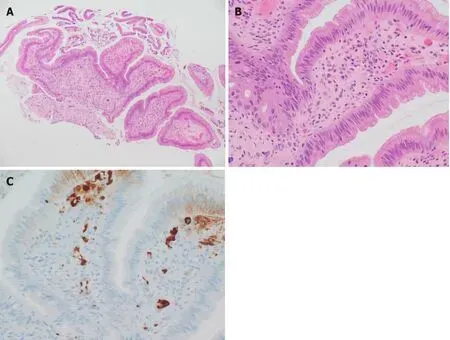
Figure 2 Histological assessment of biopsy specimen. A: Biopsy of the ampulla of Vater showed inflammatory cell infiltration and granulation tissue[hematoxylin and eosin (H&E) stain, × 100 magnification]; B: Many plasma cells were observed in the subepithelial stroma (H&E stain, × 400 magnification); C:Immunohistochemical staining showed a large number of IgG4 positive cells (brown, × 400 magnification).
CONCLUSION
We report a case of IgG4-SC that met the 2019 ACR/EULAR IgG4-RD classification criteria and was successfully treated using a GC with an immunosuppressant and made approved as imaging follow-up.
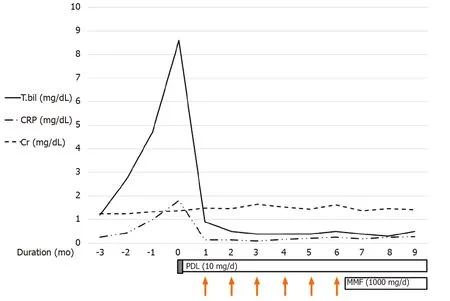
Figure 3 The initial laboratory findings indicated obstructive-pattern jaundice. Bilirubinemia (8.6 mg/dL) and a high C reactive protein (CRP) level(1.81 mg/dL) were noted. We applied high-dose systemic glucocorticoid (GC) for 3 d (prednisolone 0.5 mg/kg/d). After GC pulse treatment, the bilirubinemia and elevated CRP level had improved so we decided to administer CYC pulse therapy (1000 mg/mo, 6 mo). The maintenance therapy was prednisolone (5 mg/d) and mycophenolate mofetil (1000 mg/d). CRP: C reactive protein; MMF: Mycophenolate mofetil.
ACKNOWLEDGEMENTS
The authors thanks to all colleagues involved in the management of this case.
 World Journal of Clinical Cases2021年1期
World Journal of Clinical Cases2021年1期
- World Journal of Clinical Cases的其它文章
- Endoscopic salvage treatment of histoacryl after stent application on the anastomotic leak after gastrectomy: A case report
- Residual tumor and central lymph node metastasis after thermal ablation of papillary thyroid carcinoma: A case report and review of literature
- Necessary problems in re-emergence of COVID-19
- Simultaneous bilateral acromial base fractures after staged reverse total shoulder arthroplasty: A case report
- Krukenberg tumor with concomitant ipsilateral hydronephrosis and spermatic cord metastasis in a man: A case report
- Intraparenchymal hemorrhage after surgical decompression of an epencephalon arachnoid cyst: A case report