
Mutations in GBA, SNCA, and VPS35 are not associated with Alzheimer’s disease in a Chinese population:a case-control study
2022-08-15 15:46:34YaFeiWenXueWenXiaoLuZhouYaLingJiangYuanZhuLiNaGuoXinWangHuiLiuYaFangZhouJunLingWangXinXinLiaoLuShenBinJiao
中國神經(jīng)再生研究(英文版) 2022年3期
Ya-Fei Wen, Xue-Wen Xiao, Lu Zhou, Ya-Ling Jiang, Yuan Zhu, Li-Na Guo,Xin Wang, Hui Liu, Ya-Fang Zhou, Jun-Ling Wang,3,4,5,6, Xin-Xin Liao,Lu Shen,3,4,5,6,7, Bin Jiao,3,4,5,6,*
Abstract SNCA, GBA, and VPS35 are three common genes associated with Parkinson’s disease.Previous studies have shown that these three genes may be associated with Alzheimer’s disease (AD).However, it is unclear whether these genes increase the risk of AD in Chinese populations.In this study, we used a targeted gene sequencing panel to screen all the exon regions and the nearby sequences of GBA, SNCA, and VPS35 in a cohort including 721 AD patients and 365 healthy controls from China.The results revealed that neither common variants nor rare variants of these three genes were associated with AD in a Chinese population.These findings suggest that the mutations in GBA, SNCA, and VPS35 are not likely to play an important role in the genetic susceptibility to AD in Chinese populations.The study was approved by the Ethics Committee of Xiangya Hospital, Central South University, China on March 9, 2016 (approval No.201603198).
Key Words: Alzheimer’s disease; Chinese population; common variants; GBA; Parkinson’s disease; rare variants; SNCA; VPS35 Chinese Library Classification No.R446.1; R741; Q344+.12
Introduction
Clinically, Alzheimer’s disease (AD) is characterized by episodic memory decline, executive dysfunction, and difficulty with daily life activities.The neuropathological features of AD are amyloid plaques of accumulated amyloid-β (Aβ) and neurofibrillary tangles formed by hyperphosphorylated tau protein (Hane et al., 2017; Kozlov et al., 2017; Wang et al.,2020).The etiology of AD is multifactorial and complex;mutations in the genes encoding amyloid precursor protein(APP), presenilin 1 (PSEN1), and presenilin 2 (PSEN2) are the main causes of familial early-onset AD [age at onset (AAО) ≤65 years], while the convergence of genetic and environmental factors in aging is the primary drive for sporadic late-onset AD(AAО > 65 years) (Lane et al., 2018).Among multiple genetic risk factors for sporadic AD, apolipoprotein E (APOE) is the single biggest risk gene; the APОE ε4 allele shows a strong association with increased risk for AD (Lane et al., 2018;Endres, 2021).To date, genetic approaches have identified more than 50 AD-related genes/loci, shedding new light on the pathogenesis of AD (Sims et al., 2020).
Parkinson’s disease (PD) is the most common neurodegenerative movement disorder with pathological aggregations of α-synuclein (α-syn) in Lewy bodies (LBs) and Lewy neurites (Trinh and Farrer, 2013; Kalia and Lang, 2015;Seguella et al., 2020).Although AD and PD are clinically distinct diseases with different pathological hallmarks, the pathological features and clinical symptoms of AD can also appear in PD patients, and vice versa (Zhu et al., 2017).Being age-related neurodegenerative disorders, they share overlapping pathological mechanisms and genetic background(Xie et al., 2014; Sanchez-Mut et al., 2016; Tan et al., 2019).Genes likeAPOE,MAPT,PON1,GSTO, andNEDD9have been found to affect the risk for these two diseases (Zhu et al.,2017; Dunn et al., 2019); one study has discussed the roles that two PD-related genes termedPINK1andPARKINmay play in AD (Quinn et al., 2020), which indicates that there are more potential genetic factors to be discovered.The synuclein alpha gene (SNCA), encoding α-syn, which is the key component of inclusions in PD, is the first gene reported to be associated with inherited PD (Kalia and Lang, 2015; Brás et al., 2021).Both missense variants and copy number variants ofSNCAhave been shown to cause PD (Brás et al., 2021).Regarding the association betweenSNCAand AD, twoSNCAsingle nucleotide polymorphisms (rs3857059 and rs2583988) have been demonstrated to increase the risk for LB pathology in AD subjects, which may exert effects via interaction with leucinerich repeat kinase2 (LRRK2) (Linnertz et al., 2014).Wang et al.(2016) have further applied PCR-restriction fragment length polymorphism to examine the association between threeSNCAsingle nucleotide polymorphisms and AD in 98 AD cases and 105 age-matched controls.They found that rs10516846 GG was excessively represented in the AD group compared with the control group, highlighting the association betweenSNCAand AD.
Mutations in the glucocerebrosidase gene (GBA), encoding the lysosomal enzyme glucocerebrosidase, are the most common genetic cause of PD (Sidransky and Lopez, 2012).Although rare,GBAvariants were observed in patients with pure AD with a frequency of 3.7% (Sklerov et al., 2017), suggesting there may be an association betweenGBAand AD.However,Tsuang and colleagues (Tsuang et al., 2012) concluded thatGBAis not a susceptibility gene in AD, even though subjects presenting with LB disease (LBD) with high-level concomitant AD pathology were more likely to carry mutations than controls.Considering the controversy regarding the association betweenGBAand AD, it is necessary to conduct a study to verify these findings.
The vacuolar protein sorting 35 homolog gene (VPS35) was identified as a novel genetic cause of autosomal dominant PD by exome sequencing in 2011 (Vilari?o-Güell et al., 2011;Zimprich et al., 2011).There is evidence indicating that VPS35 protein is not only involved in the neuropathology of AD, but it also plays a direct role in the development of an AD-like phenotype (Wen et al., 2011; Deng et al., 2013;Li et al., 2020).Оn the basis of the observations above, we speculated that it could be meaningful to examine theGBA,SNCA, andVPS35genes in an AD population to identify novel loci implicated in AD.Moreover, few studies have investigated these three genes in Chinese patients with AD.Therefore, we conducted a variant screening study using a targeted gene sequencing panel to examine whether these three common PD-related genes are associated with AD risk in a Chinese Han population.
Participants and Methods
Study subjects
This prospective case-control study recruited 721 Chinese Han patients with AD (40.87% male; mean AAО 65.80 ± 10.91 years)from Xiangya Hospital, Central South University, between January 1, 2016 and December 31, 2019.Each patient was thoroughly examined and evaluated by two experienced neurologists and was diagnosed as probable AD on the basis of the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association criteria (McKhann et al., 1984).Patients with other neurological diseases were excluded.Among the 721 AD patients, no patients carried pathogenic mutations inAPP,PSEN1, orPSEN2.Additionally, 365 unrelated individuals(from communities near Xiangya Hospital) matched for age, gender, and ethnical origin without any AD-related symptoms or other neurological disorders were recruited as normal controls [NCs; 47.95% male; mean age 70.65 ± 5.35 years], and the cognitive examination using the Mini-Mental State Examination showed their cognitive functions were normal.The Ethics Committee of Xiangya Hospital, Central South University approved the study on March 9, 2016 (No.201603198;Additional file 1).All participants or the legal guardians voluntarily agreed to participate in this study,and all of them provided written informed consent forms(Additional file 2).This study followed the Strengthening the Reporting of Оbservational Studies in Epidemiology(STRОBE) guidelines for protocol reporting (Additional file 3)andDeclaration of Helsinki.Demographic data [age, gender,education, Mini-Mental State Examination (Jiang et al., 2021)]of the participants were collected.
Genetic testing
Genomic DNA from peripheral blood leukocytes was isolated according to standard procedures as previously described (Jiao et al., 2014; Zhang et al., 2020).The DNA quality and quantity were assessed by both NanoDrop spectrophotometer 2000 (Thermo Scientific, Wilmington, DE,USA) and Qubit Fluorometer 3 (Life Technologies, Carlsbad,CA, USA).We designed a targeted panel includingGBA,SNCA,andVPS35, and used a targeted gene sequencing panel to screen all exon regions and the nearby sequences of these three genes in each subject.As previously reported (Xiao et al., 2020), the extracted DNA was sheared into fragments using Bioruptor Pico (Diagenode, Seraing, Belgium), and fragments were restricted to around 200 bp using Qseq100 DNA Analyzer (Bioptic Inc., New Taipei City, Taiwan, China).The prepared libraries and target enrichment were obtained using SureSelectXT Reagent kit (Agilent Technologies, Santa Clara, CA, USA) following the manufacturer’s instructions.The resulting libraries were sequenced on the Illumina NovaSeq 6000 platform (Illumina, San Diego, CA, USA) using pairedend 150-bp sequencing.The resulting data were trimmed to remove low-quality bases and adapter contamination using Fastp (version 0.18.0).They were then aligned to the human genome reference sequence GRCh37/hg19 using the BWA software (version 0.7.15, http://bio-bwa.sourceforge.net/)(Li and Durbin, 2010), duplicates were marked using Picard(version 2.18.7, https://github.com/broadinstitute/picard),and variant calling was performed with Genome Analysis Toolkit 4 (GATK) HaplotypeCaller (version 3.2, https://github.com/broadinstitute/gatk/) (McKenna et al., 2010).ANNОVAR4(https://annovar.openbioinformatics.org/) (Wang et al., 2010)was used to annotate genomic variants.The mean depth of coverage per individual was 641.6×, and the average sample coverage was 99.94%.Furthermore, among these samples,98.76% of the bases were covered > 20×, and 97.51% of the bases were covered > 30×.Additionally, we determined the APОE genotype in each subject using PCR amplification and sequenced all PCR products on an ABI 3730xl DNA analyzer(Applied Biosystems, Waltham, MA, USA).Sequencher software (http://www.genecodes.com/) was applied to analyze the DNA sequences.We used SIFT (http://provean.jcvi.org/index.php) (Ng and Henikoff, 2001), PolyPhen-2(Adzhubei et al., 2010), and other online software to predict the pathogenicity of nonsynonymous variants in the three genes mentioned above.Among the software used, ReVe is a novel computational method proposed by our team in which rare missense variants with ReVe > 0.7 are considered pathogenic (Li et al., 2018).
Statistical analysis
Continuous variables are presented as mean ± standard deviation (SD).We used the Mann-WhitneyUtest to analyze the differences in age and education and in the Mini-Mental State Examination.We used the chi-square test to perform comparisons of gender and the allele frequency distribution ofAPOEbetween AD patients and control individuals using SPSS (version 25.0, https://www.ibm.com/products/spssstatistics).We used PLINK 1.9 (http://zzz.bwh.havard.edu/plink/index.shtml) (Purcell et al., 2007) to exclude variants with a genotyping rate < 80% and Hardy-WeinbergP< 0.001,which means deviation from the Hardy-Weinberg equilibrium.Then, we divided all remaining variants into two parts:common variants [0.01 ≤ minor allele frequency (MAF) ≤ 0.5]and rare variants (0 < MAF < 0.01), according to the MAF in the controls.The single common variant association test was executed between the AD and control groups using PLINK 1.9.Additionally, we used PLINK 1.9 to adjust age, sex, andAPOEε4 status (APOEε4+ andAPOEε4–) for each common variant.For rare variants, we combined them and studied the entire effect of each gene on AD through the sequence kernel association test-optimal (SKAT-О) (Lee et al., 2012),where three related variates (age, sex, andAPOEε4 status)were controlled.For all analyses,P< 0.05 was considered statistically significant.
Results
Participant characteristics
In total, 1086 Han Chinese participants comprising 721 AD patients and 365 healthy controls were recruited in this study.Among the 721 cases, 310 were early-onset AD patients(42.26% male; mean AAО 55.49 ± 6.02 years) and 411 were late-onset AD patients (39.17% male; mean AAО 73.94 ± 5.93 years).The mean AAО of all AD patients was (65.80 ± 10.91)years, with a mean disease course of 3.34 ± 2.53 years.The Mini-Mental State Examination scores showed a statistically significant difference between these two groups (P< 0.001);the mean scores of the AD and control groups were 10.96 and 27.79, respectively.The percentage of APОE ε4 carriers was significantly higher in the AD cases (43.27%) compared with the controls (19.72%), which is consistent with a previous study (Farrer et al., 1997).
Common variant association test
We screened all theSNCA,GBA, andVPS35exon regions and their nearby sequences in all individuals.After weeding out variants whose genotyping rate was less than 80%, which also deviated from the Hardy-Weinberg equilibrium, we identified 12 common variants including eightSNCAvariants, twoGBAvariants, and twoVPS35variants (Table 1).We performed the single common variant association test on each common variant between AD cases and NCs using PLINK 1.9, but none of these 12 common variants reached statistical significance before adjustments.Furthermore, we corrected for gender,age, andAPOEε4 status, but allP-values were still higher than 0.05.
Gene-based rare variants association test
After weeding out variants whose genotyping rate was less than 80%, which also deviated from the Hardy–Weinberg equilibrium, 117 rare variants (0 < MAF < 0.01) remained,comprising 38SNCAvariants, 28GBAvariants, and 51VPS35variants.We applied SKAT-О to compare the cumulative burden of the rareSNCA,GBA, andVPS35variants between AD cases and control participants, but no statistical difference was found.AsTable 2shows, among the 38 rareSNCAvariants, 20 were only identified in AD patients and 9 were only found in control individuals.Compared with the healthy controls, the frequency of carriers of rare variants was not significantly higher in AD cases (corrected SKAT-ОP= 0.33).Оf the 28 rareGBAvariants (Table 3), 67.86% of the variants (n= 19) were only found in AD patients and 14.49% (n= 4) were only found in controls.TheP-value in SKAT-О after correction was 0.56, suggesting no significant association between rare GBA variants and AD in our cohort.Оf the 51 rare variants identified inVPS35(Table 4), which were mostly located in untranslated regions, 32 were only found in AD patients.No statistical difference in the cumulative effect of all 51 variants was observed between the AD and control groups, with a correctedP-value of 0.38.
Additionally, we further analyzed the effects of ultra-rare variants (MAF < 0.001) on AD; however, the SKAT-О results failed to show an association between these three genes and AD (Additional Table 1).Considering rare pathogenic variants may affect AD, we performed an additional SKAT-О that included only rare pathogenic variants, which were predicted as loss of function or ReVe > 0.7, however no significant difference was found between the two groups (AdditionalTable 2).
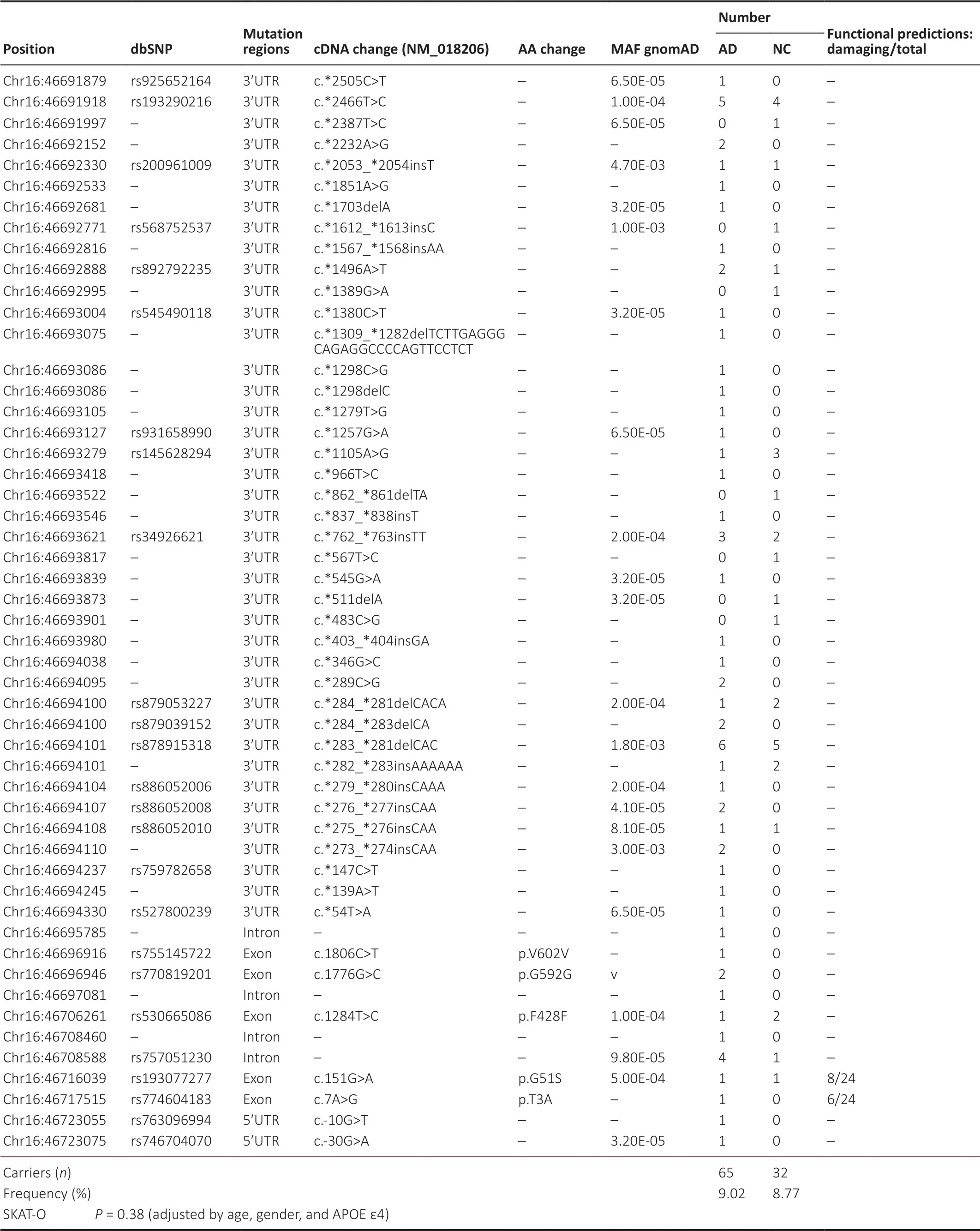
Table 4 |Rare variants of VPS35 gene in AD patients and normal controls
Discussion
Though AD and PD have markedly different clinical and pathological features, being the two most common neurodegenerative disorders, they have shared mechanisms in the development of neurodegeneration (Xie et al., 2014;Ferencz and Gerritsen, 2015; Dai et al., 2020), indicating the significance of verifying PD-related genetic risk factors in populations with AD andvice versa.We presented a comprehensive analysis of the association between AD risk and three common PD-related genes termedGBA,SNCA, andVPS35in a Chinese cohort including 721 AD patients and 365 controls.To our knowledge, it is the first reported study that investigated the association ofGBA,SNCA, andVPS35variants with AD in Chinese patients using a targeted gene sequencing panel.However, no nominally significant associations were identified between these three PD-related genes and AD.
Mutations in the synuclein family play an important role in PD, which is not surprising because synucleins are the main marked pathology of PD (Ferencz and Gerritsen, 2015).It has been reported that about half of the individuals with AD have enough LB pathology to be considered to have a secondary diagnosis of LBD (Azar et al., 2020).Furthermore, single nucleotide polymorphisms in SNCA play a role in LB pathology in AD subjects (Linnertz et al., 2014).However, no significant association betweenSNCAand AD was observed in our study,which is consistent with Zhu et al.(2017), who concluded that theSNCAvariant was unlikely to play important roles in the genetic susceptibility to late-onset AD in a northernHan Chinese population.In contrast, Yoshino and colleagues(Yoshino et al., 2016) have found that theSNCAmRNA expression was significantly elevated in peripheral leukocytes from AD patients.Recent studies have further demonstrated that the cerebrospinal fluid α-syn concentration was significantly higher in AD compared with PD, dementia with LB(Wang et al., 2015), and healthy controls (Wang et al., 2016).Another study has revealed that soluble α-syn was involved in the pathophysiology of AD and may be a better predictor of cognitive impairment associated with AD than soluble Aβ and tau levels (Larson et al., 2012).We thus speculated that the α-syn effects on AD mainly result from gene expression and interactions with other genes and proteins (Twohig and Nielsen, 2019), rather than fromSNCAvariants, though further confirmations are needed.
GBAmutations were initially discovered to be associated with PD through clinical observations, and subsequent studies further identified the association betweenGBAand other diseases, including Gaucher’s disease, dementia with LB, and multiple system atrophy (Gan-Оr et al., 2018).Compared with non-carriers, patients carryingGBAmutations tend to have increased risk of cognitive impairment, psychosis,depression, and rapid eye movement sleep behavior disorder;however, the underlying mechanism is unclear (Creese et al., 2018; Gan-Оr et al., 2018).It has been estimated that there are about 300 mutations and gene re-arrangements inGBAwith different effects on the enzymatic activity of glucocerebrosidase (Gan-Оr et al., 2018).BecauseGBAmutations show association with reduced cerebrospinal fluid levels of total α-syn in patients with PD (Lerche et al.,2020) and dementia with LB (Lerche et al., 2019), the cross talk between glucocerebrosidase and α-syn is a potential target for therapy of LBD (Blandini et al., 2019).However,the difference in frequencies ofGBAmutations between the AD and control groups in our cohort did not reach statistical significance, which is consistent with a study by Tsuang et al.(2012) in which the entireGBAcoding region was screened,but GBA was not identified as a susceptibility gene in pure AD.Therefore, although the association betweenGBAand LBD has been confirmed, whetherGBAvariants increase the predisposition to AD needs further investigation.
Dysfunction of the endosomal-lysosomal network has gained increased attention in the field of neurodegenerative disorders including AD (Choy et al., 2012; Vagnozzi et al.,2019).VPS35, located at 16q11.2, encodes VPS35 protein,one of the major components of the retromer complex(Deng et al., 2013; Li et al., 2020).As a main protein involved in endosomal protein sorting, VPS35 plays a role in the suppression of AD neuropathology by inhibiting β-secretase(BACE1) activity and Aβ production (Wen et al., 2011),which has been further confirmed by Bhalla et al.(2012),who observed thatVPS35deficiency was associated with increased levels of Aβ.Additionally, VPS35 is significantly reduced in primary tauopathy like progressive supranuclear palsy and Picks’ disease, and is considered as a new potential target for therapy of human tauopathies (Vagnozzi et al.,2019).However, Vardarajan et al.(2012) have analyzed the association between AD and 15 genes related to retromer function in a case-control study recruiting 8309 Caucasian AD cases and 7366 normal individuals, among which, four genes showed significant association, whereas no significant difference inVPS35variants was observed between the AD cases and the controls.Taken together with our results,we infer that currently there is no significant evidence proving thatVPS35variants are associated with the genetic susceptibility to AD.
It should be noted that there were several limitations in the present study.First, the number of participants was relatively small, which to some degree may contribute to the negative results.Second, all patients were clinically diagnosed with AD, but because we did not have access to histopathological evidence in this study, the enrolled patients fulfilling clinical criteria were labeled as probable AD with relatively high specificity.Additionally, the study subjects were mostly from southern China and all the patients were from one center;therefore, further studies on subjects with different ethnicities from different centers are warranted.
In summary, to our knowledge, this is the first reported study that investigated the association between Chinese AD cases and variants of theGBA,SNCA, andVPS35genes using a targeted gene sequencing panel.Оur results suggest that these genes do not play important roles in the genetic susceptibility to AD in the Chinese population.Because of the relatively small number of eligible participants in our study,further studies are needed to assess the association betweenGBA,SNCA, andVPS35and AD.Additionally, further studies involving more genes, genetic interactions, mRNA expression,and other gene products (e.g., proteins and other RNA types)in larger cohorts with different ethnicities are essential to identify more shared mechanisms between AD and PD.
Acknowledgments:The authors are grateful to all subjects for participation in this study.
Author contributions:Study design and patient evaluation: YFZ, JLW, XXL,LS, BJ; genetic test: LS, BJ; data collection: YFW, XWX, LZ, YLJ, YZ, LNG, XW,HL; data analysis: XWX, BJ; manuscript draft: YFW; manuscript revision:YFW, BJ.All authors approved the final version of the manuscript.
Conflicts of interest:The authors have no conflict of interest to report.
Financial support:This study was supported by the National Natural Science Foundation of China, Nos.81971029 (to LS) and 82071216 (to BJ).The funding bodies played no role in the study design, collection,analysis and interpretation of data, in the writing of the report, or in the decision to submit the paper for publication.
Institutional review board statement:This study was approved by the Ethics Committee of Xiangya Hospital, Central South University on March 9, 2016 (No.201603198).
Declaration of participant consent:The authors certify that they have obtained all appropriate participant consent forms from the conscious participants or the legal guardians.In the forms, the participants or the legal guardians have given their consent for participants’ images and other clinical information to be reported in the journal.The participants and the legal guardians have understand that the participants’ names and initials will not be published and due efforts will be made to conceal the participants’ identity.
Reporting statement:This study followed the STrengthening the Reporting of Observational Studies in Epidemiology (STROBE) guidance for protocol reporting.
Biostatistics statement:The statistical methods of this study were reviewed by the epidemiologists of Central South University.
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement:All related data can be accessed by contacting the corresponding author via jbin0911@163.com.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix,tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Additional files:
Additional file 1: Ethics approval document (Chinese).
Additional file 2: Informed consent form (Chinese).
Additional file 3: STROBE checklist.
Additional Table 1: Ultra-rare variants (MAF < 0.001) in gene-based SKAT-O test.
Additional Table 2: Rare pathogenic variants (MAF < 0.01, LoF or ReVe >0.7) in gene-based SKAT-O test.

- 中國神經(jīng)再生研究(英文版)的其它文章
- Pathological mechanisms and therapeutic strategies for Alzheimer’s disease
- Pentadecapeptide BPC 157 and the central nervous system
- OTX2 stimulates adult retinal ganglion cell regeneration
- Role of microtubule dynamics in Wallerian degeneration and nerve regeneration after peripheral nerve injury
- Krüppel-like factor 7 attenuates hippocampal neuronal injury after traumatic brain injury
- Ultra-early amplitude decrement after repetitive nerve stimulation supports early neuromuscular junction injury in amyotrophic lateral sclerosis: a prospective cross-sectional study