
Reactive molecular dynamics insight into the thermal decomposition mechanism of 2,6-Bis(picrylamino)-3,5-dinitropyridine
2024-04-11 03:36JianboFuHuiRenXinzhouWuYongjinChenMiZhangYazhiCheng
Defence Technology 2024年3期
Jianbo Fu, Hui Ren, Xinzhou Wu, Yongjin Chen, Mi Zhang, Yazhi Cheng
State Key Laboratory of Explosion Science and Technology, Beijing Institute of Technology, Beijing 100081, China
Keywords:PYX Thermal decomposition ReaxFF-lg MD simulations Excellent thermostable explosives
ABSTRACT 2,6-bis(picrylamino)-3,5-dinitropyridine (PYX) has excellent thermostability, which makes its thermal decomposition mechanism receive much attention.In this paper, the mechanism of PYX thermal decomposition was investigated thoroughly by the ReaxFF-lg force field combined with DFT-B3LYP(6-311++G) method.The detailed decomposition mechanism, small-molecule product evolution, and cluster evolution of PYX were mainly analyzed.In the initial stage of decomposition, the intramolecular hydrogen transfer reaction and the formation of dimerized clusters are earlier than the denitration reaction.With the progress of the reaction,one side of the bitter amino group is removed from the pyridine ring, and then the pyridine ring is cleaved.The final products produced in the thermal decomposition process are CO2, H2O, N2, and H2.Among them, H2O has the earliest generation time, and the reaction rate constant (k3) is the largest.Many clusters are formed during the decomposition of PYX, and the formation,aggregation,and decomposition of these clusters are strongly affected by temperature.At low temperatures (2500 K-2750 K), many clusters are formed.At high temperatures (2750 K-3250 K), the clusters aggregate to form larger clusters.At 3500 K, the large clusters decompose and become small.In the late stage of the reaction,H and N in the clusters escaped almost entirely,but more O was trapped in the clusters, which affected the auto-oxidation process of PYX.PYX’s initial decomposition activation energy (Ea) was calculated to be 126.58 kJ/mol.This work contributes to a theoretical understanding of PYX’s entire thermal decomposition process.
1.Introduction
Excellent thermostable explosives play a crucial role in industry and are often used in oil field development,rocket separation,and the development of new explosives formulations [1-3].2,6-bis(picrylamino)-3, 5-dinitropyridine (PYX) was first synthesized in 1972 by Coburn et al.[4].Due to its excellent thermostability(melting point 460°C, thermal decomposition peak temperature 373°C), good safety, and low production cost, PYX has attracted much attention in the field of thermostable explosives [5,6].It is gradually replacing 2,2′,4,4′,6,6′-Hexanitrostilbene (HNS) in many commercial applications where thermostable explosives are required [5,6].Tomasz et al.suggested by spectroscopic analysis that the extraordinary stability of PYX is mainly due to the presence of strong hydrogen bonding within the molecule and the lack of SP3hybridization in the carbon ring, whose significant coupling effect makes the molecule stable[7].Thomas et al.successfully prepared PYX and its salts by the improved synthesis method [5].The sensitivity test showed that the impact sensitivity of PYX was 10 J(BAM drop weight), and the friction sensitivity was 360 N (BAM drop weight), which was higher than HNS.The standard molar enthalpy of the formation of PYX calculated by the modified CBS-4M atomic basis set method is 43.7 kJ/mol.Hui et al.designed and calculated a series of derivatives of PYX at the B3LYP/6-311++G level based on density functional theory (DFT).The results show that some derivatives have better explosive properties and stability.They are expected to become new candidates for thermostable explosives [8].Currently, the research on PYX primarily focuses on the optimization of the synthesis process and the design of derivatives [5,7-10], and there are few reports on the thermal decomposition mechanism of PYX.For energetic materials,understanding the mechanism and details of thermal decomposition is very important for the safe use of energetic materials, the feasibility of guiding large-scale synthesis, the stability of long-term storage,and the sensitivity to various stimuli[11-15].
The thermal decomposition mechanism of explosives often needs to be explained from the perspective of atoms or molecules,which is precisely the shortcoming of the current experimental technology.With the improvement of computer hardware equipment, molecular simulation technology has become a powerful partner for experimental and theoretical analysis[16-20].ReaxFF,developed by Duin et al.[21],has been able to perform calculations for larger systems, simulating the breakage and generation of molecular chemical bonds with an accuracy close to that of quantum mechanical(QM)calculations.Meanwhile,the simulations are less time-consuming and costly,and statistically significant results can be obtained.However, this force field neglects the role of intermolecular London dispersion forces,leading to errors of up to 10%-15%in its prediction of molecular crystal density.Liu et al.[22]established the ReaxFF-lg force field by improving the description of the ReaxFF force field for long-range intermolecular interactions,which significantly reduced the previously existing bias.ReaxFF-lg has been successfully practiced in studying the thermal decomposition mechanism of energetic materials.Chen et al.[23] used ReaxFF-lg to simulate the thermal decomposition mechanism of HNS,whose initial thermal decomposition is mainly by bimolecular dimerization, C-NO2bond dissociation, and nitro-nitroso(NO2-ONO) isomerization.Based on this method, they also investigated the mechanism of thermal decomposition of 4,10-dinitro-2,6,8,12-tetraoxa-4,10-diazatetracyclo [5.5.0.05,9.03,11]dodecane(TEX) [24] and ε-CL-20 [25].Jiang et al.[11] investigated the decomposition mechanism of LLM-105 using ReaxFF-lg.They found that the main paths of its initial decomposition are C-NO2breaking and hydrogen transfer,and its ring structure is more prone to form macromolecular clusters through multiple bonding.Zhong et al.[26] simulated the effect of the atmosphere on the initial thermal decomposition reaction of nano-1,3,5-trinitro-1,3,5-triazinane(RDX) by ReaxFF-lg.They found that NH3, CO, NO, and NO2can promote RDX decay, and O2can prevent it at high temperatures.Lan et al.[27] used ReaxFF-lg to study the intermediate and final products during the thermal decomposition of 3-nitro-1,2,4-triazol-5-one (NTO) and identified three possible decomposition paths.Wen et al.[28] analyzed the cluster evolution of 1,3,5-triamino-2,4,6-trinitrobenzene (TATB) under different heating conditions using ReaxFF-lg.They found that the clusters formed fastest and existed for the longest time at temperatures of 2000 K-3000 K.
In this study, the thermal decomposition mechanism of PYX at constant temperatures heating of 2500 K, 2750 K, 3000 K, 3250 K,and 3500 K and programmed heating of 300 K-3500 K were investigated using the ReaxFF-lg force field and combined with the DFT method.The main initial reaction processes, intermediate products, final products, and the evolution of the clusters were analyzed.Finally, the reaction kinetic parameters of PYX were calculated.This work contributes to an in-depth understanding of the thermal decomposition process of PYX and guides its safe production and use.
2.Methodologies
2.1.Simulation details
The whole simulation was carried out in Large-scale atomic/molecular massively parallel simulator (Lammps) software [29],and the thermal decomposition mechanism of PYX was investigated using the ReaxFF-lg force field.The cell parameters of PYX were obtained from the Cambridge Crystallographic Data Centre(CCDC, number: 1,429,070) and determined experimentally by Klapotke et al.[5].The molecular structure of PYX is shown in Fig.1(a).The space group of PYX crystal is P212121, the lattice parameters are a = 14.518 ?, b = 17.661 ?, c = 18.320 ?, Z = 8, the density is 1.757 g/cm3.The unit cell model is shown in Fig.1(b).A 5×2×3 supercell was built as the initial model for the simulation by zooming in along the a, b, and c direction of the unit cell, as shown in Fig.1(c).The PYX supercell contains a total of 240 molecules (12,240 atoms).
The PYX supercell is first optimized for energy minimization.The energy minimization was performed using the Polak-Ribiˋere version of the conjugate gradient(CG)algorithm,with the stopping tolerance for energy set to 10-6and the stopping tolerance for force set to 10-9(Kcal/mol)/?.Then the Nose’-Hoover chain method was chosen to perform an isothermal-isobaric ensemble molecular dynamic (NPT-MD) simulation at 1 atm pressure and 300 K temperature for 5 ps to relax the supercell model.The initial atomic velocities are sampled according to the Maxwell-Boltzmann distribution, and the equations of motion are integrated using the velocity Verlet algorithm.Canonical ensemble molecular dynamic(NVT-MD) simulations were performed for the relaxed model at 2500 K,2750 K,3000 K,3250 K,and 3500 K for a time duration of 350 ps,respectively.During the constant temperature process,the supercell at room temperature is heated to near the target temperature in a very short period(≤0.3 ps),as shown in Table S1 in the Supplement Information.To study the cluster formation process,NVT-MD was performed at a heating rate of 8 K/ps from 300 K to 3500 K for a total of 400 ps.The time step for all simulations is set to 0.1 fs, and the information on bonds is recorded every 50 fs.Information such as potential energy (PE), temperature, pressure,atomic position,and velocity are recorded every 100 fs.The above data were collected to analyze the evolution of the PE and the number of molecular species, the evolution of small molecule products and clusters, and the path of thermal decomposition of the system during the thermal decomposition of PYX.The molecular structures were visualized by OVITO software[30].
2.2.Method to calculate the reaction kinetic parameter
The rate constant can be described by the Arrhenius equation(Eq.(1)), which calculates the activation energy and the preexponential factor of the reaction:
where T is the temperature, k is the rate constant, A is the preexponential factor, Eais the activation energy, and R is the universal gas constant.
The thermal decomposition process of energetic materials consists of three stages: the initial decomposition stage, the intermediate decomposition stage, and the final product evolution stage.The initial decomposition stage is between when the reactants begin to decompose (t0) and when PE reaches the highest point(tmax).The intermediate decomposition stage is after tmax.The rate constant k1of the initial decomposition stage of PYX thermal decomposition is obtained by fitting the change in the number of initial reactants from t0to tmaxby the first-order decay expression N(t):
where N0is the initial molecular number of the PYX reactants,and t0is the time when the PYX reactant begins to decompose.
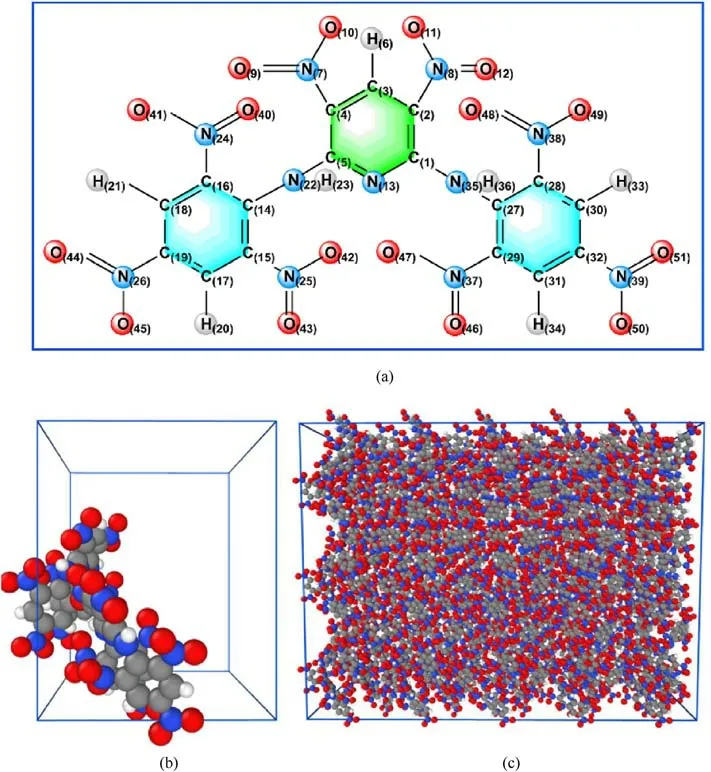
Fig.1.Structures for the PYX molecule (a), unit cell (b), and supercell (c).
The rate constant k2of the intermediate decomposition stage of PYX can be obtained by fitting the falling stage of the PE curve by the U(t) exponential function:
where U∞is the asymptotic value when PE tends to balance,ΔUexo= U(tmax) - U∞, U(tmax) is the maximum value of PE.
By fitting the change in the number of final products by the C(t)exponential function, the rate constants k3of different final products can be obtained:
where C∞is the asymptotic value at which the number of final products produced by the thermal decomposition of PYX tends to equilibrium,and tiis the time of product formation.
These computational methods have been validated in many energetic materials research works [11,23,24,31,32].
2.3.DFT calculations
In order to verify the accuracy of the ReaxFF-lg force field to optimize the PYX molecule and simulate the thermal decomposition process, Gaussian09 software [33] was used to calculate the geometric configuration and energy of the reactants,intermediates,and products in the PYX molecular decomposition process.The model is fully optimized at the B3LYP/6-311++G (d, p) level.The vibrational frequency was calculated to determine no imaginary frequency.
3.Results and discussion
3.1.Feasibility verification of ReaxFF-lg
To verify the applicability of the ReaXFF-lg force field to PYX,the cell parameters and density of the supercell model after relaxation were compared with the experimental results of Klapotke et al.as shown in Table 1.Compared with the experimental values, the maximum error of the optimized relaxation model parameters does not exceed 3.27%, which is a good fit.To improve theverification accuracy,PYX molecules’bond lengths and angles after relaxation were compared with the experimental values and DFT calculations,respectively,as shown in Table 2.The maximum errors were 9.26% and 7.20% compared to the experimental values and DFT results, respectively, from the bond length of N26-O45.A comparison between the radial distribution functions (RDF) for all the molecules in the 5×2×3 PYX supercell and the experimental values is shown in Fig.2.The RDF of the calculation results is very

Table 1 The lattice parameter of PYX.
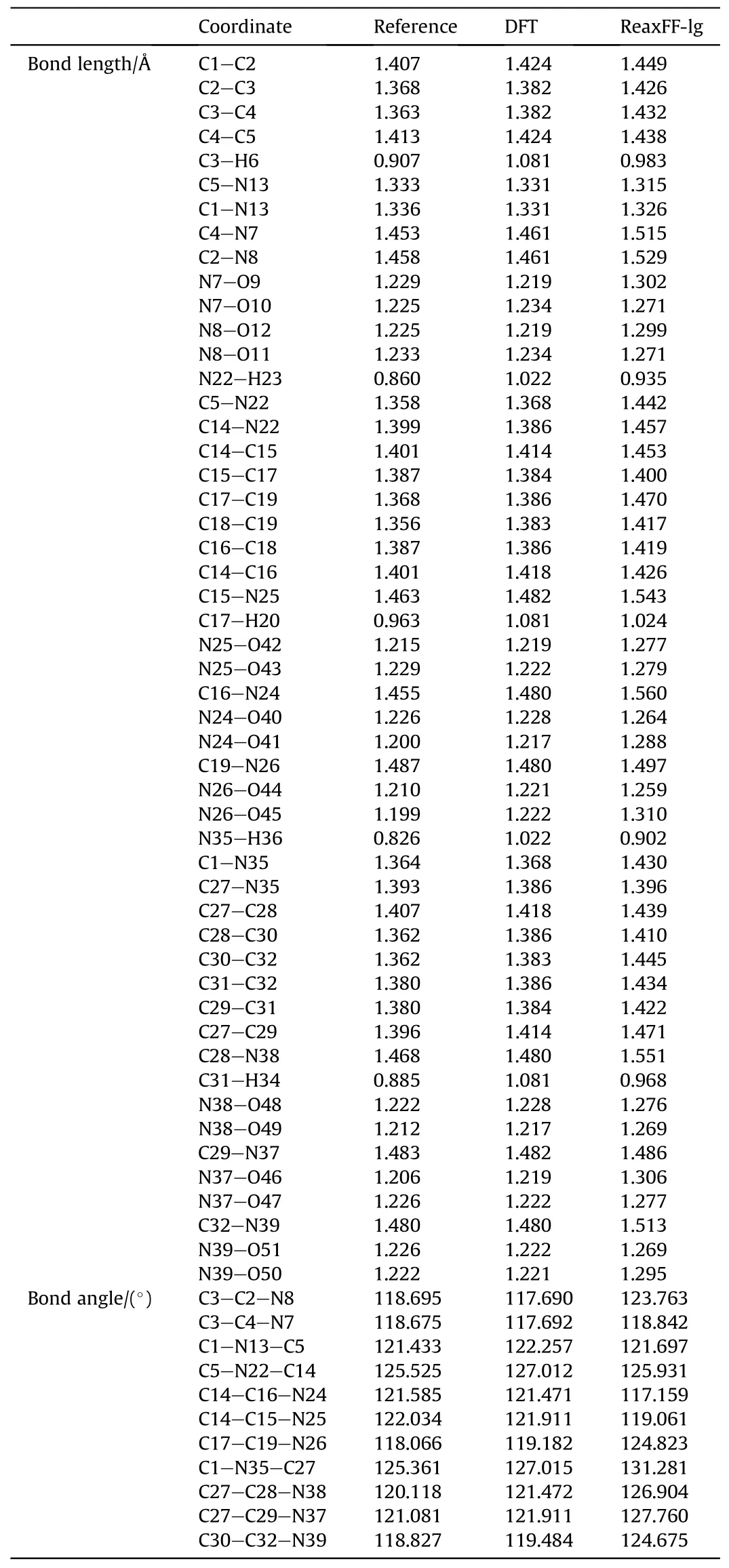
Table 2 Bond lengths and bond angles of PYX.
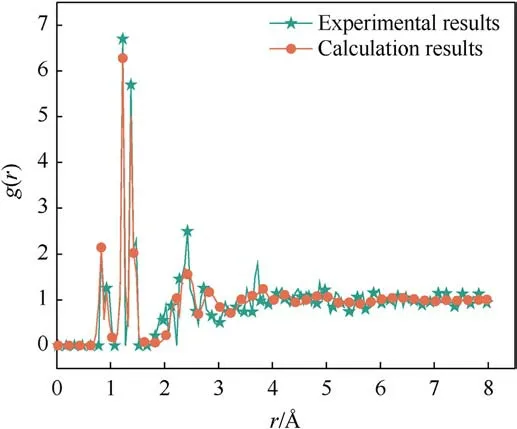
Fig.2.Radial distribution functions g(r)of the PYX crystal using the ReaxFF-lg relaxed and experimental structures.
close to the experimental RDF (Fig.2).Calculated positions of the main peaks are 0.822 ?, 0.930 ?, 1.226 ?, 1.378 ?, 1.470 ?, and 2.370 ?,whereas the main peaks obtained from experimental data are 0.830 ?, 0.945 ?,1.224 ?,1.375 ?,1.473 ?, and 2.420 ?.Therefore, the ReaxFF-lg force field can accurately predict the crystal structure of PYX.The applicability of the ReaxFF/lg force field was further verified by comparing the nitro dissociation energies of gaseous PYX molecules calculated by ReaxFF/lg and DFT methods,as shown in Table 3.The calculations show that the larger deviations of the nitro dissociation energy from ReaxFF/lg compared to DFT are 9.8%and 9.1%from C4-N7 and C2-N8,respectively,and the remaining deviations are not higher than 6%.In general, the molecular structure of PYX after optimization using the ReaXFF-lg force field is not changed, the deviation from the experimental and DFT calculated values is slight, and the accuracy is within the acceptable range.Therefore,the ReaxFF-lg force field applies to the simulation of PYX.
3.2.Evolution of potential energy and total species
The magnitude of the potential energy is mainly determined by the positions of the molecules in the system, which change as the system warms up and the potential energy fluctuates.Whether the evolution of potential energy eventually tends to equilibrium or not is an essential indication of whether the PYX thermal decomposition reaction is in equilibrium.Fig.3 shows the evolution of the potential energy with time at different temperatures(the raw data curves of the potential energy can be viewed in Fig.S1 in theSupplement Information).As seen in Fig.3,the system absorbs heat leading to a rise in potential energy,which involves PYX’s primary initial decomposition process.The maximum energy is reached and then decreases, which signals that the reaction enters the spontaneous decomposition stage.This process will generate many intermediates and high-frequency chemical collisions between intermediates to form final products.As the final products continue to increase, the potential energy stabilizes, and the thermal decomposition reaction reaches equilibrium.The time to start the decomposition of the reactants (t0) is the same for different temperatures.In addition, the completion of decomposition (tend) and the time to reach the maximum potential energy(tmax)all decrease with the increase in temperature,as shown in Fig.4.This suggests that increasing the temperature accelerates the decomposition process.Apparently,t0is generally shorter than tmax,indicating that the initial decomposition of PYX did not immediately trigger the system to enter the spontaneous decomposition stage, which is consistent with the results of Xiong et al.[34].
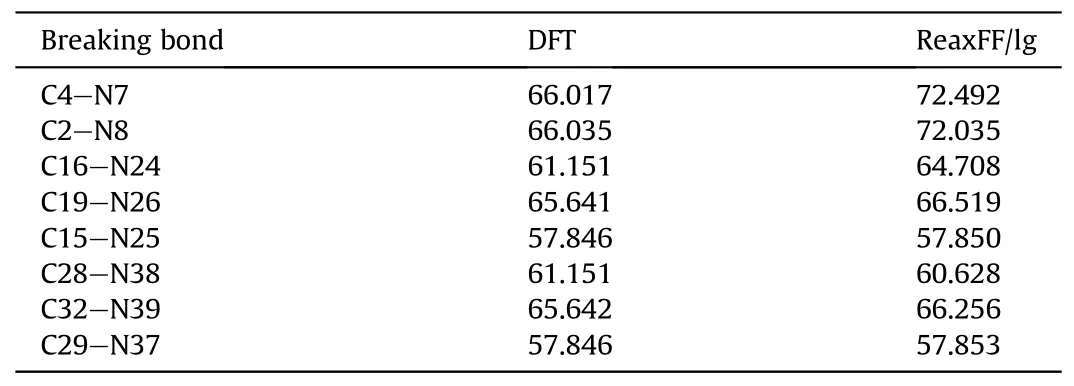
Table 3 Dissociation Barriers (in kcal/mol) of a Gas-Phase PYX Molecule.
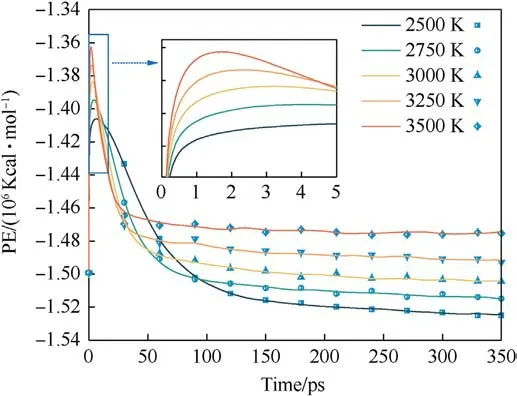
Fig.3.The evolution of potential energy at different temperatures.
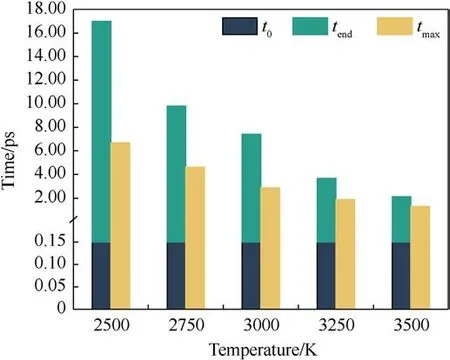
Fig.4.Key moments in the decomposition of PYX.
The evolution of the number of total molecular species in the system during the thermal decomposition of PYX with time is shown in Fig.5.As the temperature increases, the maximum number of molecular species increases, the time to reach the maximum decreases, and finally converges as the reaction proceeds.This suggests that the high temperature accelerates the decomposition of PYX while forming a wider variety of intermediates.As the reaction proceeds, the intermediate products gradually evolve into final products, causing the curve to decrease and then converge to equilibrium.
3.3.Initial decomposition pathway of PYX
The initial decomposition paths of the first 10 ps of PYX were obtained by analyzing the bond information of the decomposition process of PYX at different temperatures, as shown in Table 4.We analyzed the reactions by expunging all the reversible reactions and remaining the net ones and their frequencies.The highfrequency early decomposition reactions of PYX are shown in Fig.6 (C2-C16 reactions of PYX within 10 ps can be viewed in Table S2 in Supplement Information).
The earliest decomposition step of PYX at different temperatures is all intramolecular hydrogen transfer reactions,followed by dimer formation and denitration reactions.Dehydroxylation reaction occurs from the PYX molecule after intramolecular hydrogen transfer.It can be seen that the dissociation of the trigger bond(C-NO2) [16,23,35-39], which is often used to predict the sensitivity of energetic materials[40],does not occur in the earliest step.At 2500 K, the denitration reaction occurred at 0.25 ps, later than the intramolecular hydrogen transfer reaction and dimer formation time.Fig.7 compares the changes in the primary reaction frequencies with increasing temperature.The frequencies of intramolecular hydrogen transfer, dehydroxylation, and dimerization reactions show a decreasing trend with increasing temperature,while denitration reactions, in contrast, show an increasing trend.High-frequency intramolecular hydrogen transfer reactions occur first at low temperatures because the energy provided by low temperatures is low, and the energy barrier of these reactions is lower than that of denitration reactions.With the increase in temperature, the energy of the system increases, and the C-NO2bond begins to be destroyed more and earlier.
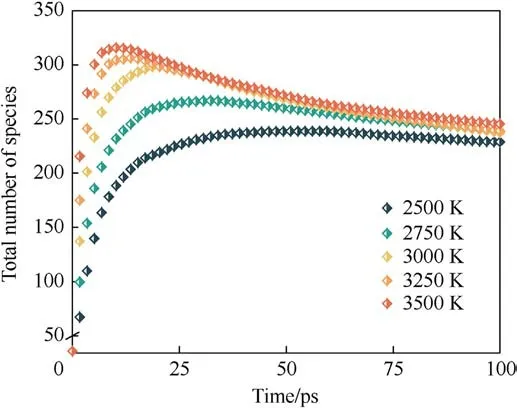
Fig.5.Evolution of total species for the system.
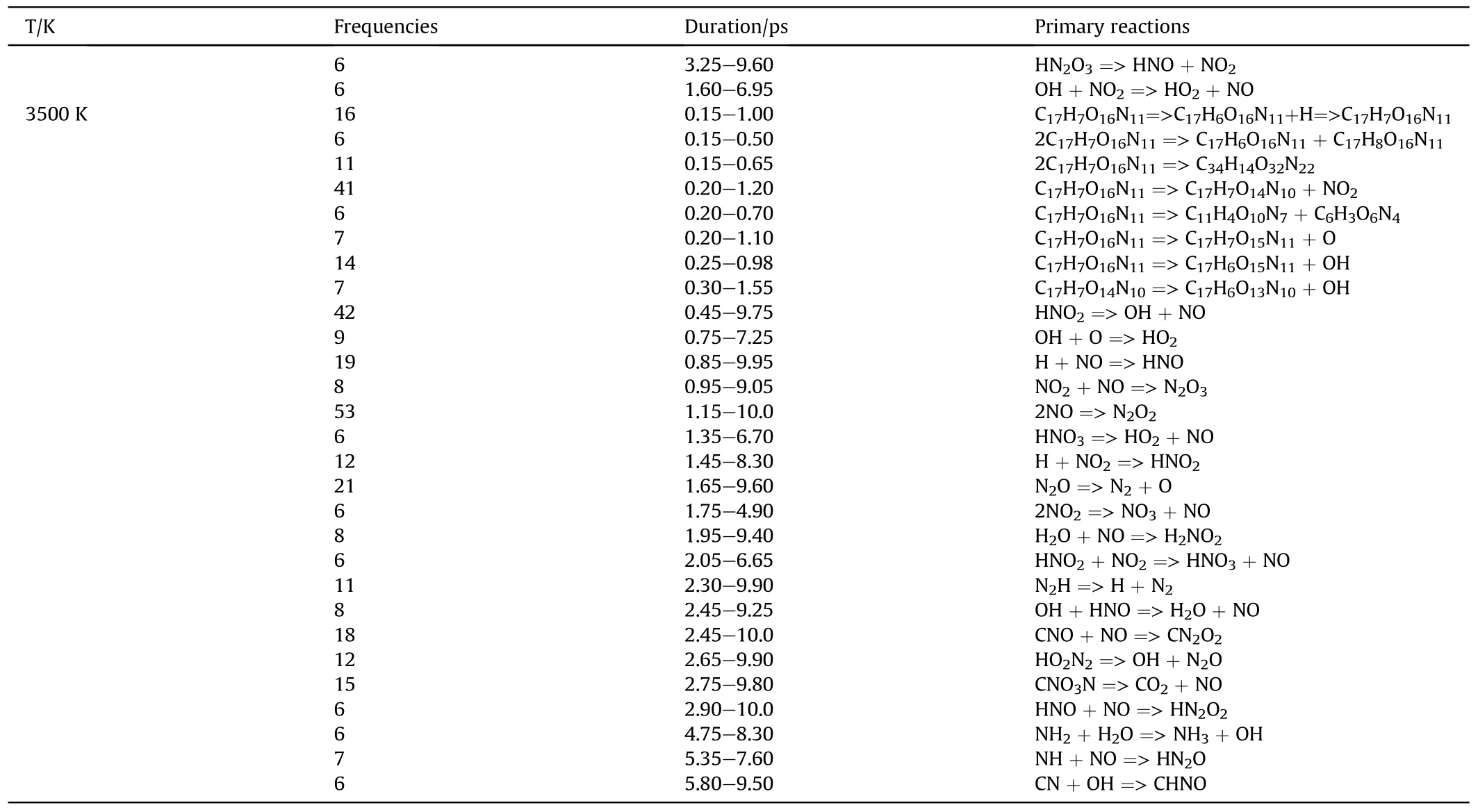
Table 4 (continued)
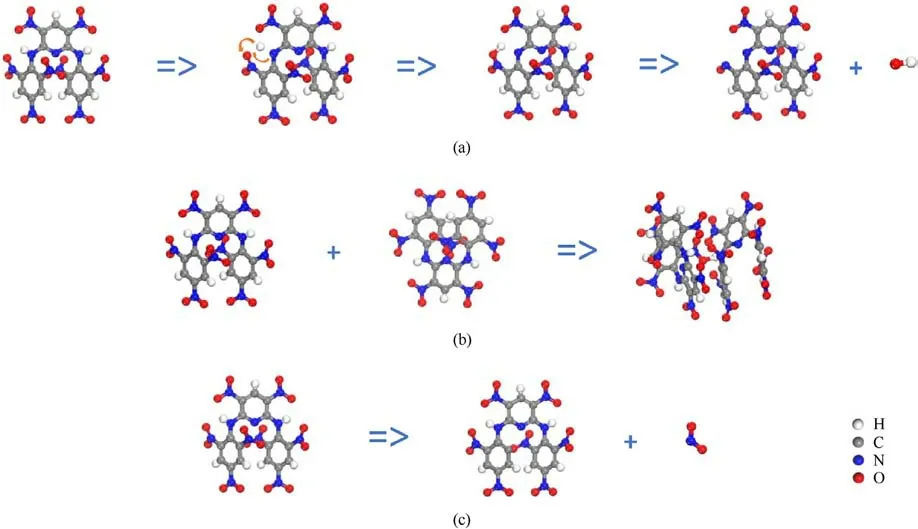
Fig.6.High-frequency initial decomposition reactions of PYX: (a) C17H7O16N11 =>C17H6O16N11 + H =>C17H7O16N11 =>C17H6O15N11 +0H; (b) C17H7O16N11 + C17H7O16N11 =>C34H14O32N22; (c) C17H7O16N11 =>C17H7O14N10 +NO2.
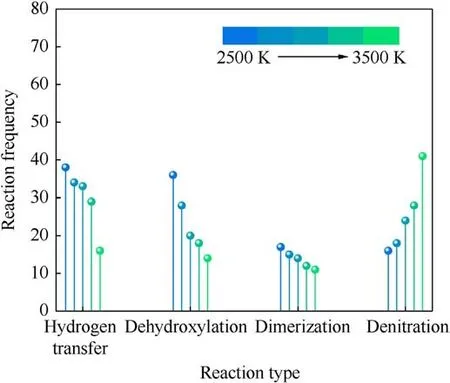
Fig.7.Comparison of the reaction frequencies of the main reactions in the early thermal decomposition of PYX.
We analyzed the bonds file (2500 K) of full simulation time(350 ps)and obtained a detailed thermal decomposition process of PYX by summarizing the reaction frequencies of all decomposition reactions and tracing the main high-frequency reactions,as shown in Fig.8.It can be seen from Fig.8 that there are three main initial decomposition paths of PYX, among which the frequency of intramolecular hydrogen transfer as the initial decomposition path is the highest.The decomposition process is divided into five main stages.In the first stage, H from the -NH- group on the PYX molecule is transferred to the adjacent -NO2group, followed by a dehydroxylation reaction.PYX molecules with intermolecular hydrogen transfer or denitration reaction as the initial decomposition path will also occur in intramolecular hydrogen transfer reaction after the initial decomposition reaction and then dehydroxylation.The second stage mainly occurs in the detachment of the nitro group and the detachment of the nitroso group.In the third stage,due to the detachment of nitro,hydroxyl,and other groups,the molecular structure of PYX loses symmetry.It becomes unstable,resulting in the detachment of the bitter amino group on the pyridine ring in the molecule.This triggered a substantial structural change in the PYX molecule.In the fourth stage,the PYX molecular structure is further disrupted, and the pyridine ring is cleaved to form small carbon-containing molecules.In the fifth stage,the small molecules formed after the cleavage of the pyridine ring collide and react with each other or combine with the large clusters formed in the system and evolve to form final products.
To validate the bias of PYX’s initial decomposition path, the three possible initial decomposition paths of the PYX molecule and the dissociation energies of all C-NO2were calculated by the DFT method at the B3LYP/6-311++G (d, p) level, as shown in Fig.9(detailed dissociation energy data for C-NO2can be viewed in Fig.S2 in the Supplement Information).The nitro dissociation energies of the different sites of PYX differed,with L3 and R3 being the smallest and M1 and M2 being the largest (From Fig.S2), with dissociation energies ranging from 57.85 kcal/mol to 66.04 kcal/mol.The energy barrier of intermolecular hydrogen transfer is 56.55 kcal/mol.It can be observed from Fig.9 that only 34.03 kcal/mol is required for the transfer of the H atom on -NH- to the adjacent nitro group,and the subsequent dehydroxylation reaction is 45.34 kcal/mol, which is all lower than the energy of the denitration and intermolecular hydrogen transfer reactions.Therefore,the initial decomposition of PYX is more likely to start from path ②.This is in general agreement with the results of ReaxFF-lg calculation.
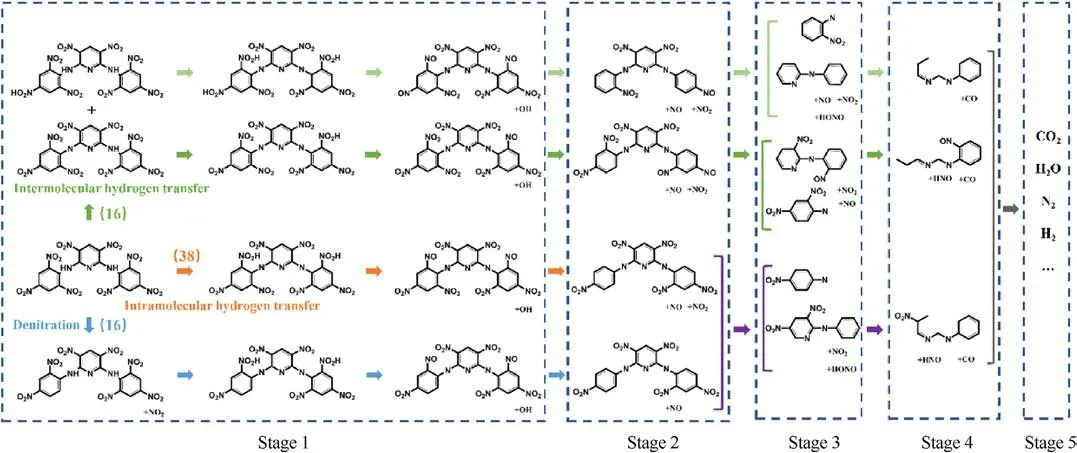
Fig.8.Thermal decomposition of PYX molecule at 2500 K.

Fig.9.Free energy barriers (ΔG) at 298 K at the theoretical level of B3LYP/6-311++G (d, p) for the two most energetically favorable paths initiating the decay of PYX: the NO2 partition and the H transfer.
3.4.Evolution of small molecule products
The main small molecules produced during the thermal decomposition of PYX are CO2, H2O, N2, H2, HONO, NO2, NO, and HNO.Among them, the first four products are stable in the late reaction stage, and the remaining small molecule products mainly appear in the early stage.The evolution law of the number of these small molecules with time at different temperatures is shown in Fig.10.The increase in temperature accelerates the time to reach equilibrium in the number of final products and the time to peak in the number of intermediate products, indicating that increasing temperature accelerated the decomposition of PYX.The final product has the highest N2and the lowest amount of H2.In the early stage of decomposition, H2O is produced first and quickly.Because the H atom in the -NH- group is transferred to the adjacent-NO2group to form a hydroxyl group,detaching from the molecule and combining with the free H to form H2O.As the temperature increases, the rate of CO2catching up with H2O is accelerated since many ring structures of PYX are opened and oxidized to form CO2earlier at high temperatures.With the decomposition of PYX, the amount of NO2quickly reached a maximum, and then NO2began to decline.This is mainly because many small molecules react to consume NO2, while NO2decomposes to produce NO.The amount of NO still increases when NO2starts to decrease, which is caused by the decomposition of HONO and HNO to generate NO and OH,and these small groups will continue to react to generate H2O and N2.
3.5.Evolution of clusters
In this paper,the products formed during the decomposition of PYX with a relative molecular weight(MW)heavier than the initial molecule(MW >621)are defined as clusters.The evolution of the number of clusters and the MW of the maximum cluster at different temperatures is shown in Fig.11.As seen in Fig.11(a),the maximum cluster number shows an evolutionary trend of increasing(2500 K-2750 K), then decreasing (2750 K-3250 K), and then increasing (3250 K-3500 K).Obviously, in the low-temperature range (2500 K-2750 K), the increase in temperature promotes the decomposition of PYX,and the collisional combination between small molecules and PYX molecules forms a large number of clusters with lower MW, resulting in a higher number of clusters.The clusters move slowly in the low temperature range(2500 K-2750 K).The probability of collisional aggregation between clusters is low,so larger clusters are not easily formed,which leads to the phenomenon that the number of clusters formed is large.However,the quality of clusters is low,as shown in Fig.11(b).As the temperature continues to increase (2750 K-3250 K), the motion of clusters with low MW is accelerated, which makes the collision frequency between clusters increase, resulting in the formation of clusters with large MW.So,the phenomenon is that the number of clusters decreases, but the maximum cluster mass increases in this temperature range.When the temperature rises to 3500 K, the high temperature causes the decomposition of large clusters to form small clusters, so the cluster mass decreases, but the number of clusters increases.
In order to better observe the evolution law of clusters, we observed the changes of clusters under heating conditions(300 K-3500 K,8 K/ps),as shown in Fig.12.Observing Fig.12,it is found that the evolution of the MW of the largest cluster with time tends to a normal distribution as the temperature increases.The cluster formation starts from the transfer of the first H atom to the PYX molecule and gradually forms a dimer (C34) to a trimer (C51),finally expanding to the largest cluster (C620).A specific range of temperature has a significant gain effect on cluster formation.As the temperature increases, the decomposition of large clusters decreases the MW.This pattern is consistent with the constant temperature process.
The evolution of the atomic ratios in the clusters at different temperatures with time is shown in Fig.13.In the early stage of cluster formation, the contents of H, O, and N are at high levels,which start to decrease until equilibrium as the reaction proceeds.The rate of decrease is accelerated with the increase in temperature.In the late stage of decomposition,the N and H in the clusters almost entirely escaped,and the O content remained at a high level.More O is trapped in the clusters, which is not favorable for the autoxidation process of PYX in the late stage of decomposition.
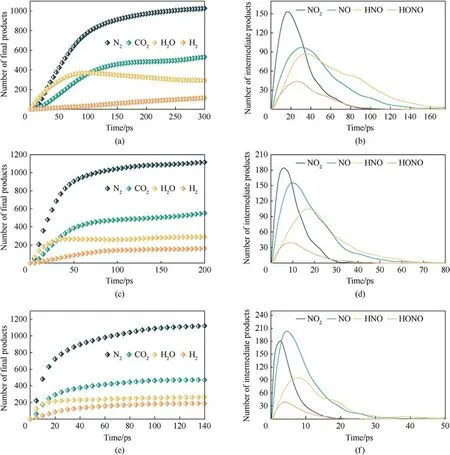
Fig.10.Evolution of the amounts of the small molecule products with time at different temperatures.
3.6.Reaction kinetic parameter analysis of PYX
Based on the study of TNT by Naomi Rom et al.[32],we divided the thermal decomposition process of PYX into three stages: the initial decomposition stage,the intermediate decomposition stage,and the final product evolution stage to solve its activation energy,the pre-exponential factor, and rate constant in this research.The reaction rate constants k1at different temperatures were obtained by fitting the curves of the variation of the initial reactant amounts with time for the initial decomposition stage (t0-tmax) during the thermal decomposition of PYX by Eq.(2)(the fitting process can be viewed in Fig.S3 in the Supplement Information), as shown in Table 5.Eq.(1) was used to fit k1.The activation energy and preexponential factor were calculated, as shown in Fig.14.The Eaof the initial decomposition stage of PYX is 126.58 kJ/mol,and ln(A)is 22.10 s-1.We compared the Ea(calculated value)of PYX with NTO(66.08 kJ/mol[27]),FOX-7(98.27 kJ/mol[41]),CL-20(95.50 kJ/mol[42]),HMX(112.60 kJ/mol[43]),and TEX(128.78 kJ/mol[24]).It is obvious that the Eaof PYX is higher than most used energetic materials, and the Eais closer to that of TEX.In the intermediate decomposition stage, the reaction rate constants k2at different temperatures were obtained by fitting the potential energy exothermic stage curve by Eq.(3)(the fitting process can be viewed in Fig.S4 in the Supplement Information),as shown in Table 6.A fit to k2was performed,as shown in Fig.15.The Eafor the intermediate decomposition stage of PYX was obtained as 113.89 kJ/mol and ln(A) as 26.20 s-1.
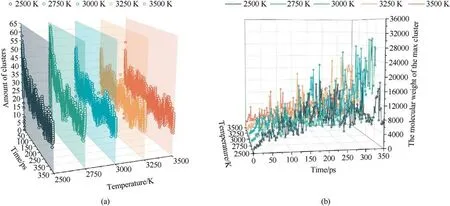
Fig.11.Evolution of number and MW of clusters with time at different temperatures.
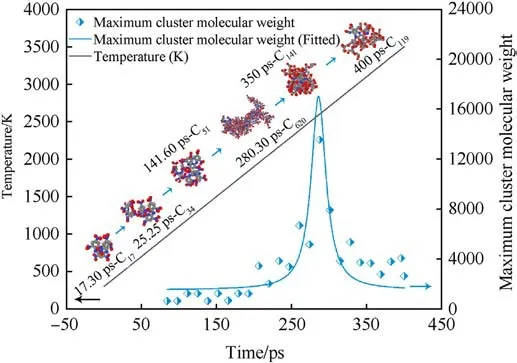
Fig.12.Cluster evolution during the temperature increase from 300 K to 3500 K.

Table 5 T0, tmax, and k1 at different temperature.
The final products from the thermal decomposition of PYX are mainly N2, H2, CO2, and H2O.The curve of the number of final products with time was fitted using Eq.(4), as shown in Fig.S5 in the Supplement Information.The variation of the rate constants with the temperature is shown in Fig.16.It can be seen from the figure that the k3of all final products increases with temperature.Among them, H2O has the largest k3, the fastest production rate,and is more influenced by the temperature increase.K3of N2,CO2,and H2increase smoothly with temperature.
4.Conclusions
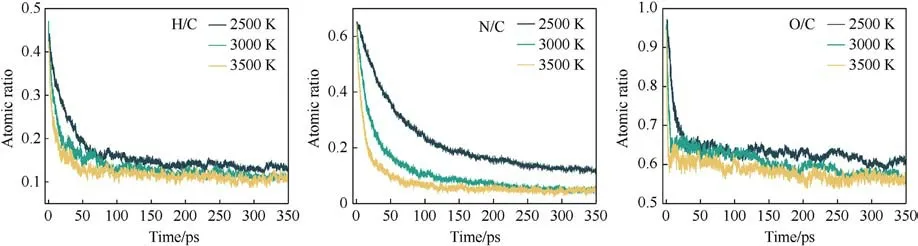
Fig.13.Atomic ratios of clusters at different temperatures.
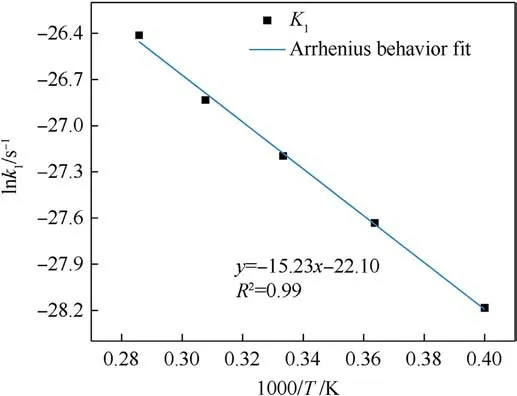
Fig.14.The logarithm of the initial reaction rate against inverse temperature from 2500 K to 3500 K.
The thermal decomposition process of PYX was simulated at different temperatures (2500 K, 2750 K, 3000 K, 3250 K, and 3500 K; 300K-3500 K, 8 ps/K) using the ReaxFF-lg force field, and the accuracy of the ReaxFF-lg was verified by the DFT method.The detailed decomposition process, cluster evolution characteristics,and final product information of PYX molecules were obtained by analyzing PYX’s reaction path, reaction frequency, and product evolution process.The initial decomposition of PYX starts with the intramolecular hydrogen transfer, denitration, intermolecular hydrogen transfer,and dimerization reactions also occur,which are critical steps in the initial decomposition.The energy barrier for intramolecular hydrogen transfer is lower than those for denitration and intermolecular hydrogen transfer reactions, confirmed by the calculated results at the B3LYP/6-311++G level.As the decomposition proceeds, the PYX structure loses symmetry and becomes unstable,and the bitter amino group is removed from the pyridine ring,followed by cleavage of the pyridine ring.During the thermal decomposition of PYX, the small molecule products generated are mainly CO2,H2O,N2,H2(final products),HONO,NO2,NO, and HNO (intermediate products), among which H2O is generated at the earliest time,and N2is generated in the enormous quantity.Many clusters were generated during the decomposition of PYX.The number of clusters showed a trend of increasing, then decreasing,and then increasing with the temperature.Because the increase in temperature favored the formation of large clusters,and the decomposition of large clusters occurred at high temperatures.In the late stage of decomposition, the clusters have low H and N contents and high O contents, which have unfavorable effects on the subsequent autoxidation process of PYX.Eafor the initial decomposition stage and the intermediate decomposition stage of PYX are 126.58 kJ/mol and 113.89 kJ/mol, respectively.In the final product evolution stage,the k3value of H2O is higher than the other final products, and the temperature has the greatest effect on it.

Fig.15.The logarithm of the middle reaction rate against inverse temperatures from 2500 K to 3500 K.
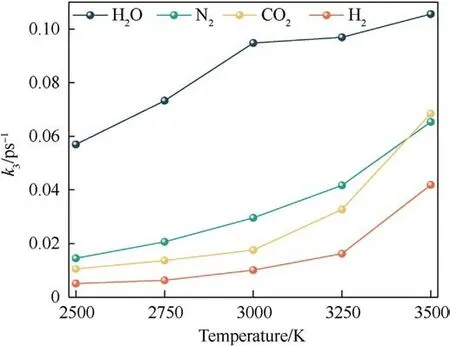
Fig.16.Relationships between the reaction rates of the final products (k3) and the temperature.
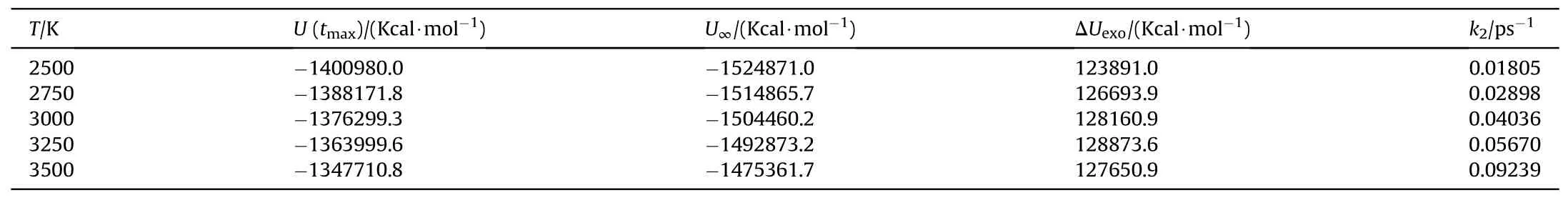
Table 6 Parameters describing the exponential behavior of the change of the PE with time.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was funded by the National Natural Science Foundation of China (Grant No.21975024).
Appendix A.Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.dt.2023.07.013.

- Defence Technology的其它文章
- Explosion resistance performance of reinforced concrete box girder coated with polyurea: Model test and numerical simulation
- An improved initial rotor position estimation method using highfrequency pulsating voltage injection for PMSM
- Target acquisition performance in the presence of JPEG image compression
- Study of relationship between motion of mechanisms in gas operated weapon and its shock absorber
- Data-driven modeling on anisotropic mechanical behavior of brain tissue with internal pressure
- The effect of reactive plasticizer on viscoelastic and mechanical properties of solid rocket propellants based on different types of HTPB resin