
Potential impact of combined inhibition of 3α-oxidoreductases and 5α-reductases on prostate cancer
2019-04-10 06:56:34MichelFindloDnielGewirthJmesMohler
Asian Journal of Urology 2019年1期
Michel V.Findlo,Dniel T.Gewirth,Jmes L.Mohler,*
aDepartment of Urology,Roswell Park Comprehensive Cancer Center,Buffalo,NY,USA
bHauptman-Woodward Medical Research Institute,Buffalo,NY,USA
KEYWORDS Androstanediol;Dihydrotestosterone;Dutasteride;3α-oxidoreductases;Androgen deprivation therapy;Abiraterone
Abstract Prostate cancer(PCa)growth and progression rely on the interaction between the androgen receptor(AR)and the testicular ligands,testosterone and dihydrotestosterone(DHT).Almost all men with advanced PCa receive androgen deprivation therapy(ADT).ADT lowers circulating testosterone levels,which impairs AR activation and leads to PCa regression.However,ADT is palliative and PCa recurs as castration-recurrent/resistant PCa(CRPC).One mechanism for PCa recurrence relies on intratumoral synthesis of DHT,which can be synthesized using the frontdoor or primary or secondary backdoor pathway.Androgen metabolism inhibitors,such as those targeting 5α-reductase,aldo-keto-reductase family member 3(AKR1C3),or cytochrome P450 17A1(CYP17A1)have either failed or produced only modest clinical outcomes.The goal of this review is to describe the therapeutic potential of combined inhibition of 5α-reductase and 3α-oxidoreductase enzymes that facilitate the terminal steps of the frontdoor and primary and secondary backdoor pathways for DHT synthesis.Inhibition of the terminal steps of the androgen metabolism pathways may be a way to overcome the shortcomings of existing androgen metabolism inhibitors and thereby delay PCa recurrence during ADT or enhance the response of CRPC to androgen axis manipulation.
1.Prostate cancer and androgen deprivation therapy
Prostate cancer(PCa)growth and recurrence relies on the interaction between the androgen receptor(AR)and testicular ligands,either testosterone(T)or dihydrotestosterone(DHT).Men with advanced PCa receive androgen deprivation therapy(ADT).ThegoalofADTistolowerlevelsofcirculating Tand impair AR activation in order to induce PCa regression.However,ADT is palliative and PCa recurs as lethal castration-recurrent/resistant PCa(CRPC)[1-5].Several mechanisms contribute to PCa persistence during ADT and the transition to CRPC,such as AR mutations,AR hypersensitivity,natural or iatrogenic-induced expression of AR splice variants[6],and intratumoral synthesis of T or DHT[7-9].Intratumoral T or DHT synthesis produces tissue T levels similar to androgen-stimulated PCa and benign prostate,and tissue DHT levels,although reduced,remain suf ficient to activate AR[7,8,10].
PCa cells have available up to three androgen metabolism pathways to produce DHT[8,11-16].The frontdoor pathway uses the two adrenal androgens,dehydroepiandrosterone(DHEA)and androstenedione(ASD),to synthesize T.DHEA and ASD are synthesized in the adrenal gland using a combination of cytochrome P450 17A1(CYP17A1)and HSD3B2 enzymes.The terminal step of the frontdoor pathway uses 5α-reductases(SRD5A1,2,3)to 5α-reduce T to DHT.The terminal steps of the primary and secondary backdoor pathways do not use T to make DHT.Instead,the primary and secondary backdoor pathways use a combination of SRD5A,CYP17A1 and 3α-oxidoreductase enzymes to produce 5αandrostane-3α,17β-diol(androstanediol)or 5α-androstane-3,17-dione(androstanedione;5α-dione).The terminal step of the primary backdoor pathway uses 3α-oxidoreductases to oxidize androstanediol to DHT.The secondary backdoor pathway uses 3α-oxidoreductases to reduce 5α-dione to DHT(Fig.1).The secondary backdoor pathway,named for its discovery after the primary backdoor pathway,is synonymous with the Sharifi“alternative” [11]and Penning “alternate” [13]backdoor pathways,and Corcoran “5α-dione”pathway[17].
The goal of this review is to describe the shortcomings of available androgen metabolism inhibitors and to discuss the therapeutic potential of combined inhibition of the 5α-reductases and 3α-oxidoreductases that catalyze the terminal steps of the frontdoor and primary and secondary backdoor pathways for DHT synthesis.
2.CYP17A1 activity,inhibitors and mechanisms of PCa resistance
The concept of inhibition of adrenal androgen production began with Huggins and Scott in 1945[18],who reported that adrenalectomy depleted testicular and adrenal androgens.However,adrenalectomy also produced adrenal insuf ficiency,which was lethal to patients without glucocorticoid replacement therapy[19,20].Since 1945,several adrenal androgen biosynthesis inhibitors were identified,such as aminoglutethimide[21,22],which impaired 11βhydroxylase,and ketoconazole[23-25],which targeted 21-and/or 11β-hydroxylase,17α-hydroxylase and 17,20-lyase activity,to deplete circulating adrenal steroid levels,such as DHEA-sulfate[24].However,aminoglutethimide and ketoconazole were not ideal inhibitors for a number of reasons including drug toxicity[26,27].
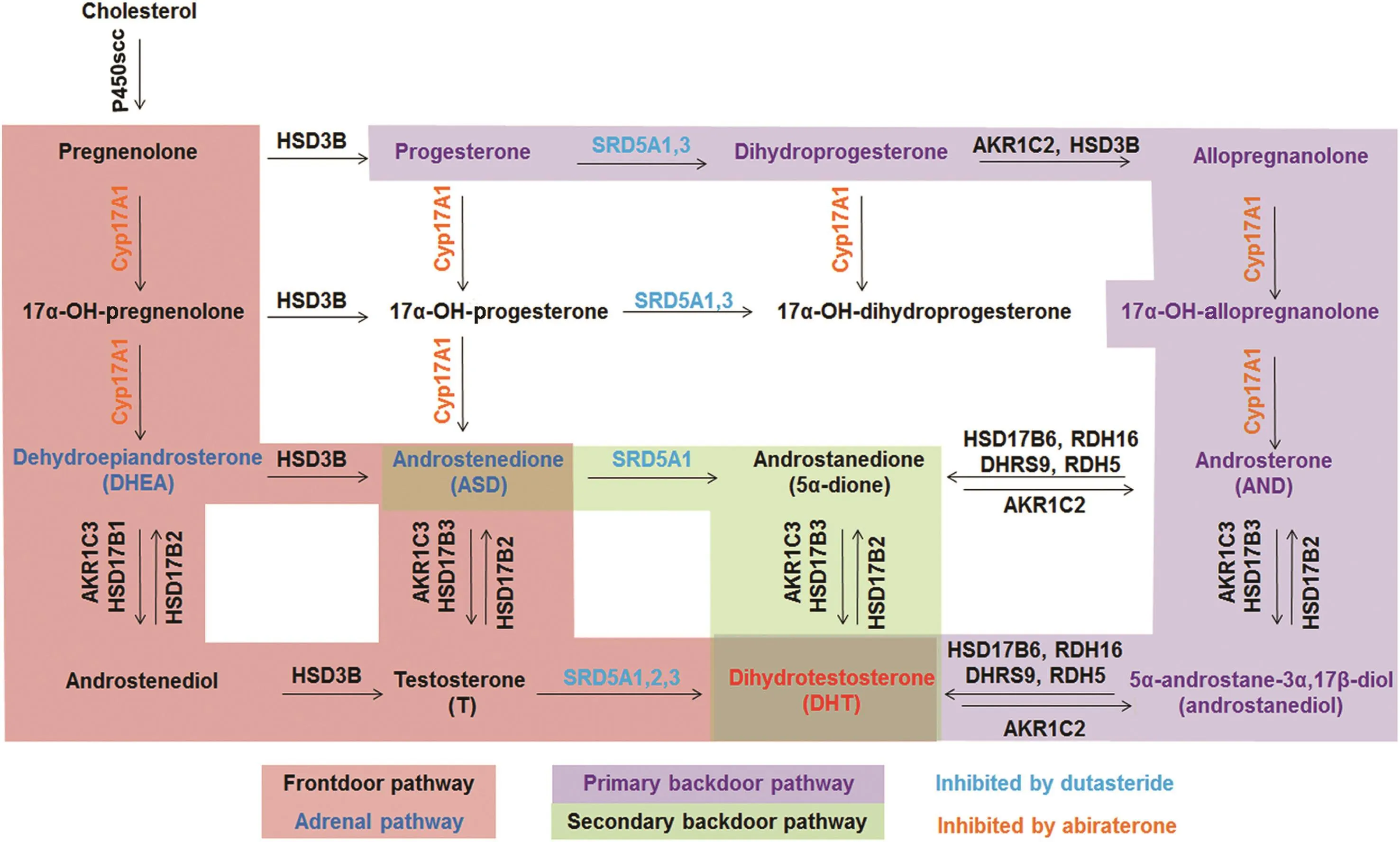
Figure 1 Pathways to DHT synthesis(modified from Ref.[62]).5α-reductase(SRD5A)1,2 or 3(frontdoor pathway;pink)metabolize testosterone to dihydrotestosterone(DHT).3α-oxidoreductase(primary backdoor pathway;purple)or aldo-keto reductase(secondary backdoor pathway;green)enzymes convert androstanediol or androstanedione,respectively to DHT.
The CYP17A1 inhibitor,abiraterone,impairs both the 17α-hydroxylase and 17,20-lyase activities of the enzyme,thus blocking adrenal androgen synthesis[28,29].Abiraterone depletes PCa or CRPC tissue androgen levels and extends patient survival approximately 4 months when used for CRPC,which garnered Food and Drug Administration(FDA)approval[30-33].However,inhibition of CYP17A1 by abiraterone can lead to glucocorticoid insufficiency and mineralocorticoid excess.Hypertension,hypokalemia and peripheral edema occur with suf ficient frequency that abiraterone is co-administered with prednisone[34-36].
The earlier clinical failure of abiraterone(and other CYP17A1 inhibitors)has been attributed to several mechanisms that involve AR splice variants[37],circulating DHEA-SO4that remains in spite of abiraterone treatment[38],increased expression of CYP17A1[39,40],or other primary[37,41]and/or secondary backdoor pathway[39]androgen metabolism enzymes.CYP17A1 inhibition induces progesterone accumulation,which competes with abiraterone for CYP17A1[42].CYP17A1 inhibition may result in up-regulation of CYP11A1 and the aldo-keto reductase family 1 member C3(AKR1C3);both enzymes are capable of increasing intra tumoral de novo androgen synthesis by activating different androgen metabolism pathways[42].Chang et al.[43]reported HSD3B1 mutations promote DHT synthesis despite therapeutic intervention.The concept of interrupting intratumoral androgen metabolism has merit,but CYP17A1 inhibition occurs too early in the DHT synthesis pathways,which enables PCa cells to alter the pathways used to produce DHT.
Therefore,inhibition of the terminal steps of the primary and secondary backdoor pathways using a combination of SRD5A and 3α-oxidoreductase enzyme inhibitors may improve PCa response.Inhibition of the steps immediately proximal to DHT synthesis will render pathway switching ineffective,which may lower DHT levels better than any androgen metabolism inhibitor used alone.
3.5α-reductase inhibition alone is insuf ficient to deplete intratumoral DHT levels
The 5α-reductases(types I-III)encoded by SRD5A1,2 or 3 play an essential role in metabolism of progestagens,glucocorticoids and androgens.These enzymes irreversibly reduce the double bond at C4 and C5 in Ring A(Fig.2;purple)[44-46]of substrates such as progesterone,cortisolor T,to produce dihydroprogesterone,dihydrocortisolor DHT,respectively[46].The type II reductase encoded by SRD5A2 is predominant in benign hyperplastic prostate and is the target of finasteride.Dutasteride inhibits type I and type II 5α-reductase,and may also inhibit type III as well[47].PCa may depend more on SRD5A1 and SRD5A3[48].Finasteride treatment enhanced the extent of response to ADTwhen initiated simultaneously with ADT[49-53],which suggests 5α-reductase inhibition may be useful when used earlier for advanced PCa.However, finasteride and dutasteride both have proven ineffective in CRPC because of variable patient response,accumulation of T that enables AR activation,and/or insuf ficient depletion of intratumoral DHT levels due to active SRD5A3 or primary or secondary backdoor pathway metabolism[54-57].
4.3α-oxidoreductases inhibition lowers intratumoral DHT levels in vitro
Redox enzymes,such as hydroxysteroid dehydrogenases(HSD),aldo-keto reductases or retinol dehydrogenases(RDH)oxidize or reduce steroid substrates[8,9,58].These enzymes are responsible for steroid metabolism,which includes glucocorticoid,mineralocorticoid and androgen synthesis or degradation[59-61].
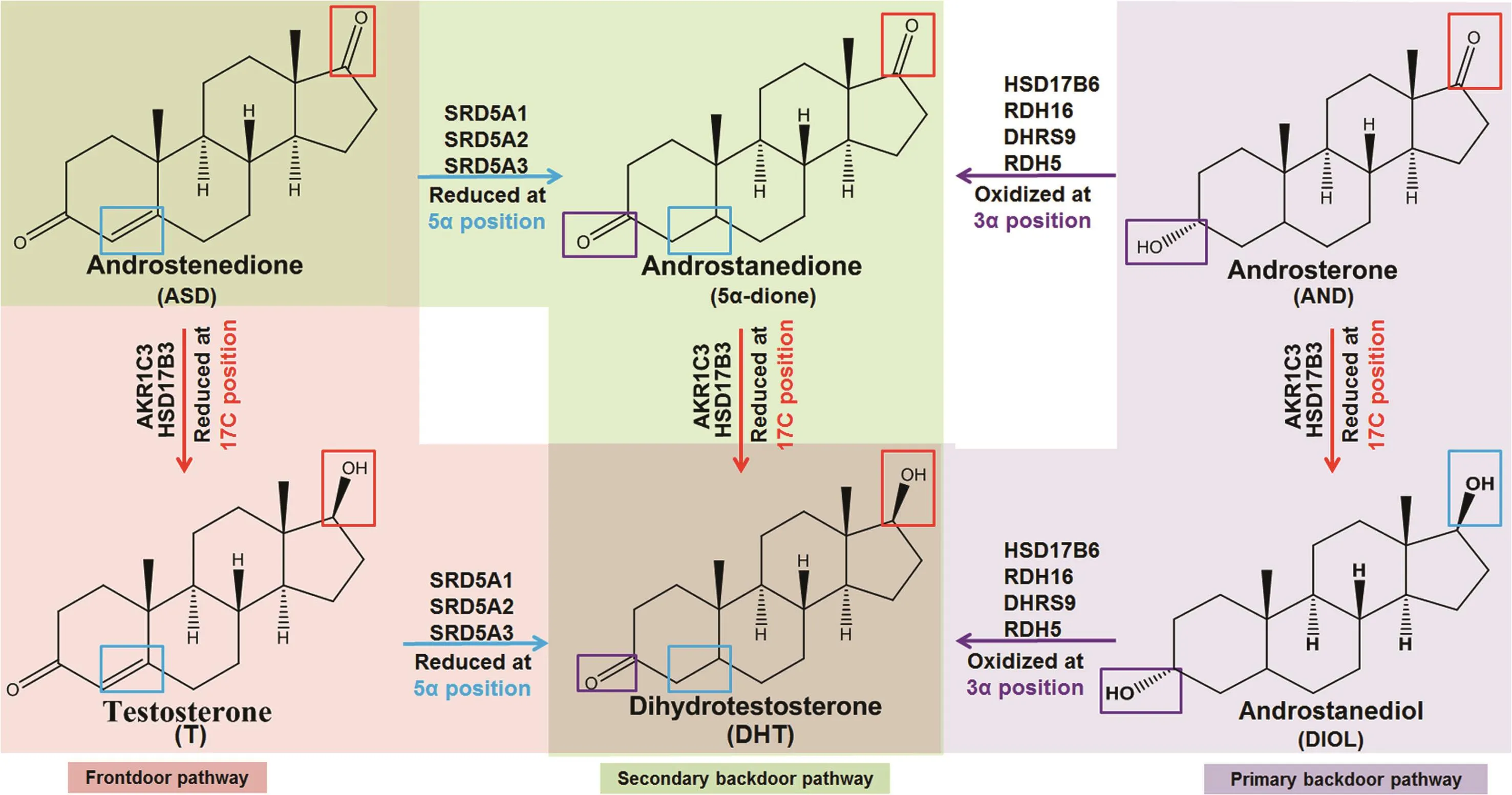
Figure 2 5α-reductase(SRD5A)(blue)and 3α-oxidoreductase(oxidation[purple];reduction[red])target sites on steroid rings.SRD5A activity(blue)occurs at the 5α position of Ring A,3α-oxidoreductase oxidation sites(purple)occur at the 3α position of Ring A and aldo-keto reduction(red)occurs at the 17β position of Ring D.
The terminal step in the primary backdoor pathway of androgen metabolism uses one of four 3α-oxidoreductases(HSD17B6;RDH5;RDH16;and dehydrogenase/reductase family member 9[DHRS9])to convert androstanediol to DHT[8,62].The 3α-oxidoreductases have conserved catalytic amino acids(Fig.3)and co-factor binding sites[63-66],and carry out similar reactions,despite having differentKmvalues[62,67].Preclinicalstudiesusing castration-recurrent CWR-R1 human xenografts demonstrated that androstanediol is converted to DHT[68].The secondary backdoor pathway requires a combination of SRD5A,to 5α-reduce ASD to 5α-dione,the four 3α-oxidoreductases,AKR1C3 or HSD17B3,to finally reduce 5α-dione to DHT[11-13,17].
A pre-clinical study using the AKR1C3 inhibitor,indomethacin,demonstrated a proof of principle that AKR1C3 inhibition overcame PCa resistance to abiraterone and enzalutamide.The study provided evidence for the necessity for identification and development of AKR1C3 inhibitors[13,69].A Phase 1/2 clinical trial that tested the ef ficacy of an AKR1C3 inhibitor,ASP9521,ended without evidence of clinical response.The authors suggested that insuf ficient PCa cell expression of AKR1C3 may have caused therapeutic failure[70].
No 3α-oxidoreductase inhibitors are used in the clinic to inhibit the four enzymes that metabolize androstanediol to DHT or 5α-dione to DHT.Recently,our group showed that combined inhibition of the four 3α-oxidoreductases and SRD5A1 or SRD5A2 lowered DHT levels better than inhibition of either enzyme group alone using in vitro PCa cell line models[62].
One challenge with using 3α-oxidoreductase inhibition therapy is enzyme redundancy.Dutasteride treatment and expression of catalytically inactive 3α-oxidoreductases impaired PCa cell line production of DHT,but did not reduce DHT levels completely in all experimental conditions[62].Expression data showed that clinical PCa specimens and PCa cell lines express at least one or more 3αoxidoreductase enzymes[62].Inhibition of one 3α-oxidoreductase enzyme may impair DHT production,but remain insuf ficient to deplete DHT levels and inactivate AR.An ideal inhibitor would target all four 3α-oxidoreductases.However,the enzymes have low protein sequence homology.This implies that there are variations in the precise substrate recognition regions of the enzymes despite their sharing100% identityamongtheircatalyticresidues[62,63].
Finally,the role of post-translational modification and activity regulation in 3α-oxidoreductase remains unclear in clinicalprostate specimens.These regulatorsof3α-oxidoreductase activity may provide an alternative target for catalytic inhibition.

Figure 3 Sequence alignment of the four 3α-oxidoreductases showed that their catalytic amino acid residues are conserved(green box).COBALT protein sequence alignment shows the conserved catalytic amino acid residues among the four 3αoxidoreductases.
5.Conclusion
The inhibitionofandrogen metabolism enzymesto improve PCa response to ADT or impair or prolong PCa transitiontoCRPC hasmerit.Despite successwith CYP17A1 inhibition(an upstream androgen metabolism enzyme),PCa recurs or CRPC persists during treatment and remains lethal[37,39-41].Inhibition of SRD5A to block the terminal step of the frontdoor pathway(conversion of T to DHT)has proven ineffective against CRPC[55-57].AKR1C3 inhibition also has been unsuccessful[70].Inhibition of the enzymes that catalyze terminal stepsin the primary backdoor androgen metabolism(conversion androstanediol to DHT)or the secondary backdoor androgen metabolism pathways(conversion androstanedione to DHT)has not been tested clinically because of a lack of candidate inhibitors.
Once inhibitors of the four 3α-oxidoreductases are developed,the lead candidate can be tested alone or combined with ADT,CYP17A1 and SRD5A1 inhibitors(Fig.4)in order to impair upstream and downstream androgen metabolism and thereby minimize the ability of PCa cells to adapt their androgen metabolism pathways during ADT.This therapy is similar to the proposed androgen annihilation therapy[71],however,this new “complete” androgen annihilation includes 3α-oxidoreductase inhibition to impair both terminal steps of the primary and secondary backdoor androgen metabolism pathways. 3α-oxidoreductase inhibition may need to be combined with anti-androgens or AR splice variant therapeutics if 3α-oxidoreductase inhibition alone reveals AR is stimulated despite inhibition of the backdoor pathway or if AR-splice variants develop.Complete androgen annihilation should improve ADT and delay or prevent death from CRPC.

Figure 4 Model of a coordinated attack on enzymes that drive dihydrotestosterone(DHT)production that should lower DHT levels better than an upstream attack of any specific enzyme.Androgen metabolism inhibitors against CYP17A1,3α-oxidoreductase,aldo-keto reductase and SRD5A used simultaneously should lower DHT levels better than individual enzyme inhibition alone.CYP17A1,cytochrome P450 17A;SRD5A,5-reductases;3α-OR,3α-oxidoreductase.
Author contributions
Study design:James L.Mohler and Michael V.Fiandalo.
Data acquisition:Michael V.Fiandalo.
Data analysis:James L.Mohler and Michael V.Fiandalo.
Drafting of manuscript:James L.Mohler,Michael V.Fiandalo and Daniel T.Gewirth.
Figure development:All figures were developed by Drs.Mohler,Fiandalo and Gewirth.
Critical revision of the manuscript:James L.Mohler,Michael V.Fiandalo and Daniel T.Gewirth.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
We thank N.Que for a careful readingof the manuscript.This study was supported by the DoD Prostate Cancer Research Program Award(No.W81XWH-16-1-0635)and the National Cancer Institute(NO.P01CA77739 and NO.R21CA205108)to JamesL.Mohler;Post-doctoralTrainingAward(No.W81XWH-15-1-0409)to Michael V.Fiandalo;DoD Synergistic Idea Development Award(No.W81XWH-14-1-0520)to Dan T.Gewirth,and NCI Cancer Center Support Grant to Roswell Park Comprehensive Cancer Center for the Bioanalytics,MetabolomicsandPharmacokinetics,PathologyNetwork,and Genomics Shared Resources(No.P30CA016056).
 Asian Journal of Urology2019年1期
Asian Journal of Urology2019年1期
- Asian Journal of Urology的其它文章
- Application of fluorescence in situ hybridization in the detection of bladder transitional-cell carcinoma:A multi-center clinical study based on Chinese population☆
- Detection of androgen receptor(AR)and AR-V7 in small cell prostate carcinoma:Diagnostic and therapeutic implications
- Albumin-linked prostate-specific antigen-activated thapsigargin-and niclosamide-based molecular grenades targeting the microenvironment in metastatic castration-resistant prostate cancer
- Prostate tumor neuroendocrine differentiation via EMT:The road less traveled
- Regulatory signaling network in the tumor microenvironment of prostate cancer bone and visceral organ metastases and the development of novel therapeutics
- Combination therapy with androgen deprivation for hormone sensitive prostate cancer:A new frontier