
Albumin-linked prostate-specific antigen-activated thapsigargin-and niclosamide-based molecular grenades targeting the microenvironment in metastatic castration-resistant prostate cancer
2019-04-10 06:56:38EmmanuelAkinboyeNathanielBrennenSamuelDenmeadeJohnIsaacs
Asian Journal of Urology 2019年1期
Emmanuel S.Akinboye,W.Nathaniel Brennen,Samuel R.Denmeade,John T.Isaacs*
Department of Oncology,Prostate Cancer Program,The Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins,The Johns Hopkins University School of Medicine,Baltimore,MD,USA
KEYWORDS Albumin-linked prodrug;Maleimide coupled albumin delivery;Thapsigargin;Niclosamide
Abstract Localized prostate cancer is curable via annihilation of the entire cancer neighborhood by surgery or local radiation.Unfortunately,once metastatic,no available therapy is curative.The vast majority will die despite aggressive systemic combinational androgenablation therapies.Thus,there is an urgent need for effective systemic therapeutics that sterilize the entire microenvironment in metastatic castration-resistant prostate cancer(mCRPC).To accomplish this goal,advantage can be taken of the unique biology of mCRPC cells.Like their normal cell of origin,mCRPCs retain expression of the prostate-specific differentiation protein,prostate-specific antigen(PSA),which they abundantly secrete into their extracellular fluid(ECF).This unique,and essentially universal,secretion of enzymatically active PSA into the ECF by mCRPCs creates an exploitable therapeutic index for activation of systemically delivered highly lipophilic toxins as “molecular grenades” covalently linked to cysteine-34 of human serum albumin(HSA)via a stable maleimide containing PSA cleavable peptide such that PSA-dependent hydrolysis(i.e., “detonation”)releases the grenades restrictively within the ECF of mCRPC.This approach decreases dose-limiting host toxicity while enhancing plasma half-life from minutes to days(i.e.,pharmacokinetic effect)and increasing the tissue concentration of the maleimide coupled albumin delivery(MAD)in the ECF at sites of cancer due to the enhanced permeability of albumin at these sites(i.e.,enhanced permeability and retention effect). This allows the MAD-PSA detonated grenades to circulate throughout the body in a non-toxic form. Only within sites of mCRPC is there a sufficiently high level of enzymatically active PSA to efficiently “pull the pin” on the grenades releasing their lipophilic cellpenetrant toxins from HSA. Thus, if a sufficient level of “detonation” occurs, this will kill mCRPC cells, and sterilize the entire PSA-rich metastatic sites via a bystander effect. In this review, two examples of such MAD-PSA detonated molecular grenades are presented-one based upon thapsigagin and the other on niclosamide.
1.Introduction
Donald S.Coffey spent more than 50 years in a quest to conquer cancer,particularly prostate cancer.Along the way,one of the most insightful challenges that Don asked each of the legion of his loyal inspired students,fellows,and faculty colleagues was whether they wanted“to study or cure cancer”.Don realized that this was a loaded question since in order to cure cancer,one needs to be armed with suf ficient basic science knowledge about cancer to acquire the “wisdom” needed to identify unique vulnerabilities and thus enable more than a simple empiric approach to the development of novel curative therapies.In addition,Don was a champion for the fact that each organ site-specific cancer presents both unique biological problems and opportunities.This realization led Don to be a strong proponent of organ site-specific basic and translational science.Based upon his inspiration and mentoring,after an initial 2 decades of“studying” prostate cancer to acquire suf ficient knowledge,our group has spent the last 2 decades translating this basic science wisdom into novel,rational therapeutic approaches.
2.Problems with metastatic castrationresistant prostate cancer(mCRPC)-Low therapeutic index for standard drugs and tumor heterogeneity
There is an urgent need to develop effective systemic therapeutics for mCRPC patients.Obviously the goal for such therapeutics is to produce death of mCRPC cells without an unacceptable amount of killing or injury to the non-malignant cells in normal host tissues.This is a daunting engineering challenge.Remarkably,modest successes,and even cures,are achieved through the use of non-selective,proliferation-dependent chemotherapeutic poisons in a limited number of cancer types due to their high proliferative rate and low celldeath threshold compared to normal tissues[1].The price for such nonselectivity,however,is dose-limiting host toxicities,particularly myelosuppression,which greatly limits therapeutic ef ficacy against the more slowly proliferating mCRPC cells.This is particularly relevant since we documented more than 20 years ago that while mCRPC cells have the lowest proliferation rate of any type of human cancer cells(i.e.,<5%of these malignant cells are proliferating per day),they are still eventually lethal since their death rate is even lower(i.e.,<3%of cells die per day)[2].Since the cells of multiple vital normal organs(e.g.,skin,bone marrow,and gut)have proliferation rates of>20%of cells per day,there is almost no therapeutic index for standard proliferation-dependent cytotoxic chemotherapy drugs for the systemic treatment of mCRPCs.Over the last 2 decades,two rapid autopsy studies,one at Johns Hopkins University[3]and the other at the University of Michigan[4]have confirmed our earlier cell kinetic data.In our study at Hopkins,the median growth fraction in 117 individual mCRPC sites was 3%and the mean was(7±1)%[3].For the Michigan study,the median growth fraction in 103 individual mCRPC sites was 5%and the mean was(5±5)%[4].These characteristic cell kinetic limitations have led to a profound re-thinking about therapeutic strategies that can activate the proliferation-independent death of mCRPC cells.
Along these lines,cancer cells often acquire an addiction to specific oncogenic signaling pathways that control both their survival and proliferation.For mCRPC,intracellular signaling by nuclear androgen receptor(AR)is the most validated of such oncogenic addictions[5,6].Thus,novel approaches to block AR signaling are being developed by many investigators in the field.While this is entirely reasonable,there are limitations to such an approach based upon the heterogeneity of AR expression within metastatic sites in individual mCRPC patients.For example,autopsy studies document that AR expression varies widely across mCRPC samples(n=265)with only 31%of tumor samples having>50%of the malignant cells expressing AR while 42%have <10%and 20%have <1%of the malignant cells expressing AR[4].Overall,this translates to a median of only 20%of the mCRPC cells expressing AR with a mean of(28±34)%[4].These results make the sobering prediction that any treatment which completely inhibits intracellular signaling by nuclear AR will be clinically useful,but probably not curative for the vast majority of mCRPC patients.
3.Rationale for prostate-specific antigen(PSA)targeting of mCRPC
To overcome the curative limitation due to the heterogeneity of AR expression,advantage can be taken of the unique biology of prostate cancer cells.Like their normal cell of origin,prostate cancer cells retain expression of the prostate-specific differentiation protein,PSA,which they abundantly secrete into their extracellular fluid(ECF)[7].Autopsy studies document that within 265 samples of metastatic sites from 30 mCRPC patients,none lacked detectable expression of PSA and in half of the samples;at least 50%of the cancer cells express PSA with a median expression of 39%and a mean of(43±35)%[4].PSA is a 33 kD single-chain glycoprotein that is synthesized and uniquely secreted in large quantities by prostate epithelial cells(i.e.,expression is nearly 3 log orders higher in prostate than any other tissue in the human body)[8].PSA is a serine protease with chymotrypsin-like substrate specificity that is found in 100 μmol/L concentrations in the seminal plasma where its major proteolytic substrates are the seminal gel-forming proteins,semenogelin I and II[9].In collaboration with Dr.Hans Lilja,we used the PSA cleavage map for the semenogelins to identified a peptide with the amino acid sequence His-Ser-Ser-Lys-Leu-Gln-//-X(HSSKLQ//X)in which hydrolysis between the carboxyl group of Gln(Q)and the amine of the C-terminal amino acid(i.e.,Q//X)is highly restricted to PSA recognition(i.e.,PSA>2 logs better hydrolysis rate for this substrate than chymotrypsin or any other known cancer-associated proteinasesincluding elastase,matrix metalloproteinase(MMP)-2,MMP-9,urokinase,plasmin,tissue plasminogen activator,thrombin,human kallikrein 1(HK1),human kallikrien 2(HK2),and fibroblast activation protein(FAP)[9].We used this HSSKLQ//X substrate to document that prostate cancer cells secrete extremely large (i.e.,1.6-2.1 μmol/L)amounts of enzymatically active PSA into their ECF[7].Once in the ECF,PSA eventually enters the blood.However,in the blood,PSA is enzymatically inactivated via covalent binding to the serum protease inhibitors α-1-antichymotrypsin and α-2-macroglobulin,which are present in vast molar(i.e.,mmol/L)excess[9].Importantly,a high level of enzymatically active PSA continues to be secreted into the ECF by prostate cancer cells even in patients with high Gleason grade and/or late-stage mCRPC patients[10].In fact,it is the continuous increase in the serum level of α-1-antichymotrypsin covalently bound PSA(i.e., “complexed” PSA),which requires enzymatically active PSA,that is used as one of the main markers of clinical progression in such lethal patients[10].Along these lines,a study from MD Anderson documented that<3%of metastatic patients(i.e.,100 out of 4145)treated for prostate cancer had disease progression without an elevation in complexed PSA in the serum[11].
The essentially universal secretion of enzymatically active PSA into the ECF by mCRPC cells creates an exploitable therapeutic index for activation of a combination ofsystemically delivered “moleculargrenades”designed so that their hydrolysis(i.e., “detonation”)releases potent lipophilic toxins(i.e.,grenades)restrictively within the ECF of mCRPC sites.The chemical engineering requirements for such a combinational PSA-detonated molecular grenade platform is that the individual toxins must be:1)highly cell penetrant(i.e.,lipophilic)when liberated,2)a potent(i.e.,nmol/L)inhibitor of vital,but complementary,intracellular targets(e.g.,endoplasmic reticulum[ER]and mitochondria),whose inhibition induces the proliferation-independent death by all cell types,and 3)capable of peptide bond formation with the C-terminal carboxyl group of Gln(Q)in the HSSKLQ//PSA peptide to form a prodrug(i.e.,molecular grenade)that is not toxic until hydrolyzed by PSA.Thus,following systemic dosing,such PSA-detonated molecular grenades circulate throughout the body in an inactive state,but only within sites of mCRPC is there a suf ficiently high level of enzymatically active PSA to ef ficiently “pull the pin” on the grenades releasing the cell penetrant toxins from the peptide.Since this hydrolysis occurs in the ECF,the toxins once liberated rapidly penetrate all cell types in the immediate vicinity due to their lipophilicity and do not reenter systemic circulation.Thus,if a suf ficient level of“detonation” occurs,this will not only kill the mCRPC cells,but sterilize the entire PSA-rich metastatic sites including all of the tumor-in filrating host cells which are stimulating the lethal cancer growth(i.e.,tumor endothelial cells,cancer associated fibroblasts[CAFs]and immunosuppressive mescenhymal stem cells[MSCs],myloid derived suppressor cells [MDSCs], and regulatory T-cells[Tregs])[12-14].An additional advantage of such selective extracellular hydrolysis is that only a fraction of the cells within the tumor microenvironment need to express PSA since its continuous enzymatic activity amplifies the level of cell penetrant toxin liberated in the ECF within the metastatic site.This stoichiometric amplification negates the problem of tumor cell heterogeneity by inducing a substantial“bystander effect” in which,like a detonated grenade,all cells within the tumor site including both malignant and in filtrating host supporting cells are killed,including those that do not express PSA.Thus,development of resistance is retarded without simultaneously producing non-selective toxicity within normal tissue sites.In addition,unlike many other therapeutic strategies in which expression of the molecular target is assumed,but not validated individually,clinical use of such PSA detonated molecular grenades is “personalized” based upon the detection of α-1-antichymotrypsin bound PSA target in the patient’s serum,thereby,validating this precision medicine approach.
4.First generation PSA-activated thapsigargin(TG)molecular grenade
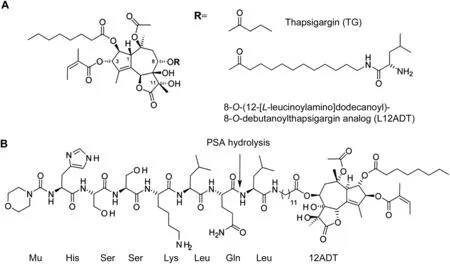
Figure 1 Chemical structures of thapsigargin(TG)and TG prodrug.(A)Structures of TG and L12ADT;(B)Structure of Mu-HSSKLQ//L12ADT.The arrow indicates the site of prostate-specific antigen(PSA)hydrolysis.
As the first example of this innovative molecular grenade strategy,a chemically-modified analog of the naturally occurring sesquiterpene γ-lactone,TG was chosen.TG(Fig.1A)isolated from the Thapsia garganica plant is highly lipophilic and thus cell permeable[15].Once inside the cell,TG is a potent inhibitor(IC5010 nmol/L)of the ER calcium ATPase(i.e.,ATP2A2 a.k.a.SERCA 2b)pumps[16].Our group initially discovered that sustained exposure of human mCRPC cells to TG induces rapid inhibition of their SERCA pumps,resulting in the depletion of the high(i.e.,>500 μmol/L)Ca2+in their ER,inducing both an ER stress response and a “capacitance entrance” of extracellular Ca2+[17].This leads to an initial increase in the intracellular free Ca2+(Cai)from~40 to>300 nmol/L within minutes followed by a return to baseline over 9-12 h,which is then followed by a delayed sustained increase in Caito>1 μmol/L over the next 18-36 h[18].The combination of ER stress and a sustained elevation of Caieventually results within several days in the apoptotic death of mCRPC with a lethal dose which causes 50%cell death(LD50)of<50 nmol/L[17-20].As part of the death response,TG treatment rapidly depletes AR protein expression in AR-positive mCRPC cells[20].Importantly,unlike drugs like doxorubicin or taxanes which are much more effective in killing proliferating cells,TG is equally effective in killing both non-proliferating and proliferating cells[20].Furthermore,TG even kills multidrug resistant cells lacking apoptotic Bak and Bax proteins[21].Such potent TG-induced killing,however,is not cancer cell specific(i.e.,TG kills normal cells equally well)[19].In particular,TG at<100 nmol/L not only kills human umbilicalvascularendothelialcells(HUVECs),but also inhibits sprouting by the surviving cells needed for tumor angiogenesis,as documented using a 3-dimensional HUVEC sprouting assay as we have described previously[22].Thus,if targeted to sites of mCRPC,TG is an ideal agent to annihilate all the cell types present within the cancer neighborhood(i.e.,microenvironment).
To target TG’s cell proliferation-independent toxicity to cancer neighborhoods,our group has collaborated with Dr.S?ren Br?gger Christensen,one of the original discoverers of TG[15].Initially,we used an iterative medicinal chemistry approach in which individual side chains of the TG molecule were selectively modified.We determined that position 8 of TG could be modified without significantly affecting SERCA binding.Based upon a combination of enzyme assays,protein crystallography,and in vitro ef ficacy,we determined that a primary amine-containing 8-O-(12-aminododecanoyl)-8-O-debutanoyl thapsigargin analog(12ADT)has an optimal-length side chain for inhibitory binding to the SERCA pump(IC5010 nmol/L)and is only slightly less lipophilic than TG,and thus has a cytotoxic potency only slightly less(LD5055±12 nmol/L)than TG(LD5013±3 nmol/L)[20,23,24].The advantage of 12ADT,however,is that it can be covalently coupled via a peptide bond between its primary amine to the C-terminal carboxylic acid of Q in the HSSKLQ PSA-hydrolysable peptide.Direct coupling,however,resulted in a conjugate that is not hydrolyzed ef ficiently by PSA[19].Subsequently,we discovered that 12ADT coupled to the C-terminal of leucine(L)to produce L12ADT(Fig.1A)is more lipophilic and thus more potent(LD5014±8 nmol/L)than 12ADT.In addition,when the N-terminus of leucine in L12ADT is coupled to the C-terminus of Q in the PSA peptide,the resulting HSSKLQ//L12ADT peptide conjugate(Fig.1B)is ef ficiently hydrolyzed(i.e.,kcat/Kmratio of 22)by PSA to liberate L12ADT[19].Thus,this HSSKLQ//L12ADT peptide conjugate is stable in human plasma and selectively toxic(LD5074±3 nmol/L)to PSA-producing prostate cancer cells in vitro[19].
Pharmacokinetic/pharmacodynamic(PK/PD)and tumor ef ficacy analyses of human mCRPC xenografts in nude mice documented that this first-generation peptide conjugation(i.e.,molecular grenade)functions to solubilize the lipophilicity of TG to allow aqueous systemic intravenous(IV)delivery of the peptide conjugate,and to decrease its penetration into cells at sites lacking enzymatic PSA,thus limiting host toxicity[19].In fact,the maximum tolerated dose(MTD)for this first generation peptide conjugation was shifted from <1 μmol/kg for TG to >12 μmol/kg.A single IV dose of 12 μmol/kg resulted in a peak serum concentration of 15 ± 1 μmol/L and a half-life(t1/2)of approximately 3 h.Over 24 h,less than 0.5%of the administered peptide-drug conjugate was observed in plasma in the free(i.e.,toxic)L12ADT form.When mice bearing growing human mCRPC xenografts were IV dosed daily for 3 days with 12 μmol/kg,the level of liberated L12ADT in the cancer tissues was 170±58 nmol/L,which is 13 times higher than its in vitro LD50[19].A regimen of five daily IV injections with 12 μmol/kg dose every 2 weeks resulted in >75%inhibition of the growth of human mCRPC xenografts in mice over a 40 day period without substantial host toxicity,but had no effect on non-PSA producing human renal carcinoma xenografts[19].However,the therapeutic index for this first generation compound is narrower than anticipated from the in vitro studies.While multiple daily doses of 12 μmol/kg are tolerated,a single dose of 36 μmol/kg is lethal to mice.Similar non-specific toxicity is observed when this first generation PSA detonated TG molecular grenade was evaluated in a preclinical cynomolgus monkey toxicology screen.These studies also documented unexpected dose-limiting toxicity.We recently discovered the mechanism for this toxicity is due to the inability of the 6 amino acid PSA carrier peptide to suf ficiently neutralize the high lipophilicity ofTG,which allowsnon-selective permeation of the non-hydrolyzed grenade into normal cells in noncancerous tissues.This result is consistent with our protein crystallization studies,which documented that once inside a cell,potent(i.e.,nmol/L)binding to and thus inhibition of the SERCA pump occurs even if the aminododecanoyl side chain in position 8 of TG is coupled to small peptides[25].
5.Rationale for maleimide coupled albumin delivery(MAD)for PSA-activated TG molecular grenade
To overcome the problem with the first generation TG-molecular grenade’s promiscuous cellular uptake,we are taking advantage of the fact that human serum albumin(HSA)has a single reactive free cysteine(cys-34)whose reactivity is so ef ficient that it has been used to produce albumin-coupled prodrugs for clinical trials[26].Based upon this realization,a second generation molecular grenade has been designed in which cys-34 of HSA is covalently bound to a “stabilized” maleimide (i.e.,2- fluoro-5-maleimidobenzoate)linker covalently coupled to the N-terminus of the PSA-activated peptide ending in L12ADT,(Fig.2).As we have reported previously,using 2- fluoro-5-maleimidobenzoate for sulfhydryl coupling to HSA produces a much more stable succinamic acid thioether linkage than compared to the more commonly used N-alkyl maleimides that produce a succinimide thioether ring linkage which undergoessignificantspontaneous cleavage by a retro-Michael reaction under physiologic conditions[27].When this spontaneous succinimide thioether ring cleavage reaction occurs in vivo,it results in dose-limiting systemic toxicity as the therapeutic agent is spontaneously released to circulate in the blood to form adducts with other sulfhydryl containing species like gluthathione,cysteine,etc.;thereby,generating a systemically toxic molecule.In contrast,opening of the succinimide-thioetherring (succinimidylhydrolysis)to form the succinamic acid thioether results in the generation of a stable drug-protein conjugation with a very long half-life of up to 2 years[27].
Using this specific 2- fluoro-5-maleimidobenzoate strategy for production of a“stabilized”MAD-conjugated second generation PSA detonated grenade accomplishes four goals.First,coupling to HSA prevents non-selective cell permeation to minimize side effects(i.e.,toxicity)to non-PSA producing host tissues outside of sites of mCRPC.Second,this coupling greatly increases the serum half-life of the second generation grenade from hours to days(pharmacokinetic effect).Third,blood vessels in sites of cancer are much more permeable to albumin than in normal tissues,and thus coupling to albumin enhances the extracellular concentration of the grenade specifically at sites of mCRPC via a tumor site-specific process known as the enhanced permeability and retention(EPR)effect[28].Fourth,this MAD approach creates a modular platform for the production of a “family” of PSA-detonated molecular grenades attacking different,but complementary,critical intracellular targets.
6.MAD for PSA-activated nicosamide molecular grenade
While this MAD approach for second generation PSA-activated TG molecular grenades should significantly increase its anti-prostate cancer ef ficacy,it is unknown whether it will be curative when used as monotherapy.What is clear,however,is that this general MAD approach provides a modular platform for the creation of other PSA-detonated molecular grenades to induce proliferationindependent cell death selectively within sites of mCRPC via complementary,but SERCA pump inhibition independent,intracellular targets.As a complementary target,we have focused on the unique vulnerability of mCRPC cells via their dependence upon “parasitic” co-option of nutrients from their supporting tumor stroma.This occurs because despite activation of tumor angiogenesis,the metabolic needs of the cancer cells eventually exceed the nutrient level provided by the tumor blood supply,producing a compromised tumor microenvironment.Under this stress,the in filtrating stroma cells composed of endothelial cells,macrophages, fibroblasts, and adiptocytes undergoes autophagy/mitophagy to liberate energy-rich nutrients like fatty acids and metabolic intermediates like glutamine and lactic acid into the tumor ECF[29].These metabolites are ef ficiently parasitized from the ECF due to enhanced expression of the specific plasma membrane transports for these substrates by the mCRPC cells[30-32].In contrast,expression of the plasma membrane glucose transporters is characteristically low in mCRPC cells consistent with their low rate of glucose uptake[33,34].These results provide a mechanistic explanation for why mCRPC cells characteristically have a low rate of glycolysis and instead derive the majorityoftheirATPgenerationfrom mitochondrial oxidation using a combination of free fatty acid,lactate/pyruvate,and glutamine/glutamate as substrates[35-37].For these reasons,[18F]- fluorodeoxy glucose(FDG)positron emission tomography(PET)imaging is typically not used in prostate cancer patients due to the frequent presence of FDG-negative metastaticsites.Thisparasiticnutrient cooption of mCRPC is thus unique compared to other solid malignancies since it is not characteristically driven by a high level of aerobic glycolysis(i.e.,lacks the Warburg effect),but instead shunts glucose carbons toward lipogenesis[36].This is also consistent with why prostate cancer cells have a striking increase in the number and pleomorphism of mitochondria[38-40].
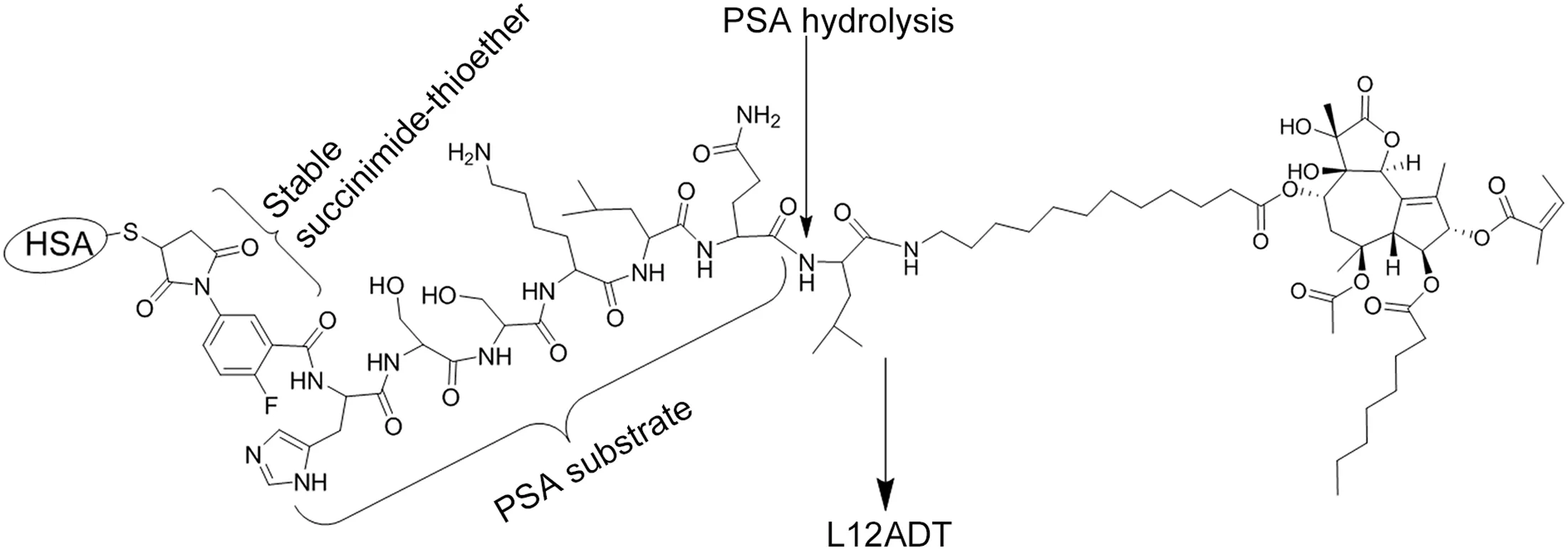
Figure 2 Structure of prostate-specific antigen(PSA)activated prodrug of 8-O-(12-[L-leucinoylamino]dodecanoyl)-8-O-debutanoylthapsigargin analog(L12ADT)with 2- fluoro-5-maleimidobenzamide covalently coupled to the N-terminal of PSA substrate histidine-serine-serine-lysine-leucine-glutamic acid(HSSKLQ).
These results support the hypothesis that additional PSA-detonated grenades targeted at inhibiting mitochondria function provides a highly rational target based upon mCRPC cells’unique dependence on “parasitic” nutrient co-option,particularly when combined with ER-targeted PSA-detonated TG grenades.Thus,we are synthesizing a mitochondrial-targeted PSA-detonated grenade based upon the FDA-approved salicylanilide anti-helminthic,niclosamide(Fig.3A).This lipophilic cell-penetrant compound was originally approved for human use 50 years ago based upon its ability to uncouple oxidative phosphorylation and stimulate ATPase activity in the mitochondria of cestodes(e.g.,tapeworms),thus killing these parasites[41].Subsequently,over the last 5 years,niclosamide had been documented to kill mammalian cells,including mCRPC cells,with an LC50<1 μmol/L[41,42].The mechanism for such cell death includes its ability to collapse the outer mitochondrial membrane potential;thereby,uncoupling oxidative phosphorylation[43-45].This uncoupling is due to niclosamide’s high lipophilicity and thus cell penetration,where it enters mitochondria to collapse the proton gradient needed for ATP synthesis[46].This protonophoric effect is dependent upon the pKaof the aromatic hydroxyl group of niclosamide,which at physiologic pH allows cycling between a neutral and anionic form(Fig.3A).This is critical since it enables delocalization of the negative charge of the anionic form to maintain its lipophilicity to allow it to cross the outer mitochondrial membrane where the proton dissociates,releasing the neutral form which re-crosses the outer mitochondrial membrane;thus collapsing the proton gradient[45,46].
Using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide(MTT)growth and clonogenic survival assays combined with in situ cell staining,we have confirmed that niclosamide not only kills mCRPC cells(i.e.,LD50is 600nmol/LforLNCaP,800nmol/LforCRW22-Rv1,750 nmol/L for PC-3,and 500 nmol/L for LAPC4 human prostate cancer lines),but that this toxicity involves loss of the mitochondrial outer membrane potential detected as a decrease in mitochondrial ability to convert JC-1 dye from a green to a red fluorescence[47].The well-established mitochondrial uncoupler, carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP),wasused for comparison(Fig.3B).We have also confirmed that when given at 25 mg/kg per day(i.e.,76 μmol/kg per day)for 2 weeks via intraperitoneal injection in 5%Tween-80/5%ethanol/90%phosphate buffered saline(i.e.,a non FDA-approved vehicle),niclosamide inhibits the in vivo growth of human mCRPC xenografts in nude mice as reported by Alan Gao’s group(Fig.3C)[42].This inhibition is due to increased cell death within the site of cancer(Fig.4).
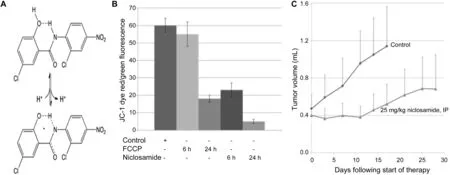
Figure 3 (A)Structure of the anionic vs.neutral form of niclosamide;(B)Loss of mitochondrial outer membrane potential detected as a decrease in JC-1 dye red/green fluorescence ratio;(C)In vivo growth inhibition of the CWR-22 human prostate cancer xenograft in nude mice(n=10 per group)by niclosamide given at 25 mg/kg per day(i.e.,76 μmol/kg per day)for 2 weeks via intraperitoneal(IP)injection in 5%Tween-80/5%ethanol/90%phosphate buffered saline.FCCP,4-(tri fluoromethoxy)phenylhydrazone.
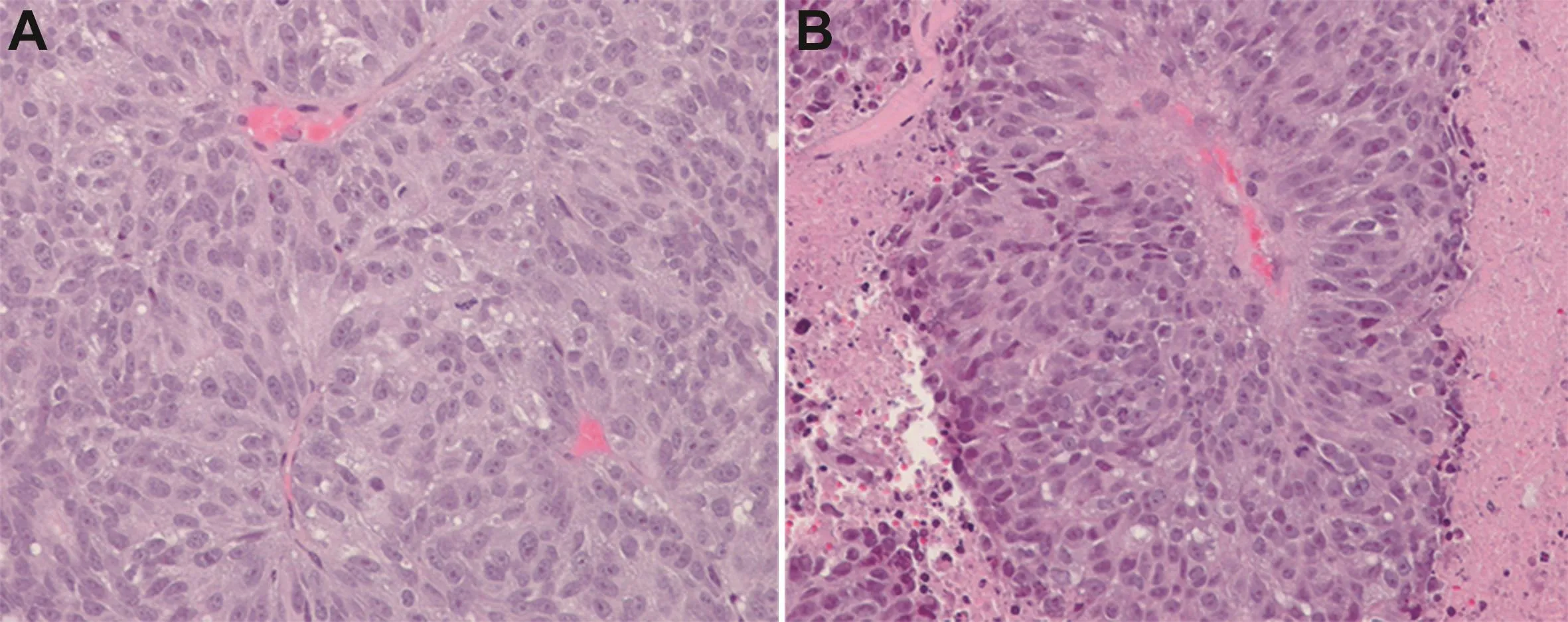
Figure 4 Histology of the CWR-22 human prostate cancer xenograft in vehical control(A)vs.niclosamide(B)treated hosts.
Niclosamide’s potent killing ability is not restricted to malignant cells,but also occurs in normal cells including endothelial cells(i.e.,LD50for HUVECs is 500 nmol/L).In addition,using a three-dimensional sprouting assay,as we have described previously[22,48],we documented that niclosamide inhibits HUVEC sprouting with an IC50of 200 nmol/L.This raises the serious issue of the therapeutic index for niclosamide as a cancer drug.In addition,a major problem for the clinical development of niclosamide for cancertherapy isitspooraqueoussolubility (i.e.,0.23 μg/mL or only 700 nmol/L according to the Merck Index).This is significant as the LD50value for killing cells is essentially equal to niclosamide’s water solubility.This makes achieving a therapeutic blood level via IV dosing using FDA-approved vehicles very difficult.To overcome this solubility issue,we coupled a glycylproline (GP)dipeptide(compound 1)to the amine of a self-cleaving linker(SCL),and theresultingcompound 2isthen coupled to the critical aromatic hydroxyl group of niclosamide(compound 3)via an ether or carbamate linkage to produce compound 4[GP-SCL-niclosamide),chemically as described in Fig.5.
Formation of GP-SCL-niclosamide(compound 4)not only solubilizes niclosamide(i.e., >100 μmol/L)but also prevents anion formation of the aromatic hydroxyl group blocking its protonophoric ability,totally preventing its protonophoric killing ability.In addition,the GP dipeptide-SCL-niclosamide is chosen because it is an excellent substrate for C-terminal cleavage between P and the selfcleaving linker by either dipeptidyl peptidase IV(DPPIV)or FAP expressed within the cancer microenvironment on the plasma membrane of prostate cancer cells[49]or tumor-in filtrating CAFs/MSCs,respectively,as we have documented previously[50-52].Once compound 4 is hydrolyzed,the liberated SCL-coupled compound undergoes spontaneous self-cleavage;as we have documented previously with other SCL coupled compounds[50,53],liberating niclosamide with its aromatic hydroxyl group now is able to ionize and thus function as cell penetrant protonophore(Fig.6).This is documented by the observation that GPSCL-niclosamide is so ef ficiently hydrolyzed by DPPIV that it has an essentially identical LD50as niclosamide for killing DPPIV expressing mCRPC cells in vitro(600 nmol/L for LNCaP,800 nmol/L for CRW22-Rv1,and 500 nmol/L for LAPC4 human prostate cancer lines),but no ability to kill normal prostatic basal epithelial cells,which lack DPPIV expression,even at 100 μmol/L.
To further restrict the killing ability of a niclosamide molecular grenade to only sites of mCRPC,we are taking advantage of two additional modifications to compound 4.First,the N-terminal G of compound 4 is being covalently linked to the C-terminal Q in the PSA peptide to produce HSSKLQ//GP-SCL-niclosamide.Since both DPPIV and FAP are endopeptidases and thus require a free N-terminal of G in order to hydrolyze the bond between P and the self cleaving linker,liberation of niclosamide from HSSKLQ//GP-SCL-niclosamide requires sequential dual hydrolysis by PSA then DDPIV/FAP.Second,as with the thapsigargin approach,the N-terminus of the HSSKLQ//GP-SCL-niclosamide is being covalently linked to cys-34 of HSA via a “stabilized” maleimide (i.e., 2- fluoro-5-maleimidobenzoate)containing linker to produce an HSA-coupled PSA/DPPIV dual-detonated niclosamide grenade(Fig.7).This is being tested for its systemic anticancer ef ficacy alone and in combination with the HSA-coupled PSA-detonated TG grenade when given intravenously in an FDA-approved vehicle(i.e.,saline).
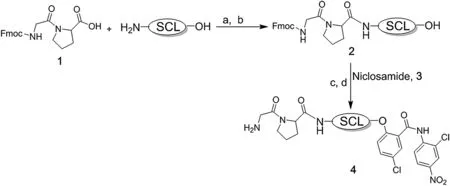
Figure 5 Synthetic scheme for glycylproline(GP)coupling via a self cleaving linkers(SCL)to the aromatic hydroxyl group of niclosamide to produce GP-SCL-niclosamide(i.e.,compound 4).SCL,self cleaving linker;a,2-Ethoxy-1-ethoxycarbonyl-1,2-dihydroquinoline(EEDQ),Dichloromethane N,N-dimethylformamide-dichloromethane(DMF-DCM);b,25%piperidine in DMF;c,Diisopropyl azodicarboxylate(DIAD),triphenylphosphine(PPh3),tetrahydrofuran(THF);d,20%piperidine/DMF.
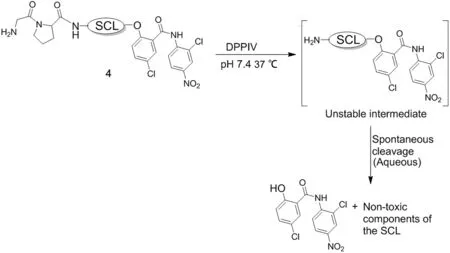
Figure 6 Schematic description of dipeptidyl peptidase IV(DPPIV)/ fibroblast activation protein(FAP)activation of compound 4 to liberate niclosamide.SCL,self-cleaving linker.
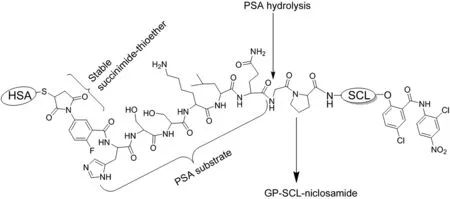
Figure 7 Schematic description of the albumin-linked dual prostate-specific antigen(PSA)/dipeptidyl peptidase IV(DPPIV)-activated niclosamide molecular grenade.HSA,human serum albumin;GP,glycylproline;SCL,self-cleaving linker.
7.Conclusion
While Don Coffey consistently championed the need for continuing expansion of basic science discoveries,he always challenged us to translate basic science discoveries into “cures” for prostate cancer.Along these lines,the rationale for several MAD-linked PSA-activated molecular grenades for targeting metastatic CRPC have been presented.Additional approaches developed by our group using PSA to active recombinant bacterial toxins(i.e.,topsalysin)and a prostate specific membrane antigen activated TG prodrug(i.e.,mipsagargin)have entered clinical testing[23,54].Similar approaches exploiting the unique enzymactic activity of other tumor-associated proteases such as FAP are being developed[52].
Author contributions
Study concept and design:Emmanuel S.Akinboye and John T.Isaacs.
Data acquisition:Emmanuel S.Akinboye and John T.Isaacs.Data analysis:Emmanuel S.Akinboye and John T.Isaacs.Drafting of manuscript:Emmanuel S.Akinboye and John T.Isaacs.
Critical revision of the manuscript:W.Nathaniel Brennen and Samuel R.Denmeade.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgments
We would like to acknowledge the Prostate Cancer Foundation,Department of Defense Prostate Cancer Research Program(W81XWH-16-1-0410),NIH Prostate SPORE(P50 CA058236),Patrick C.Walsh Prostate Cancer Research Fund,and the Hopkins-Allegheny Health Network Cancer Research Fund.Also we wish to thanks the Cell Imaging Facility,Animal Core Facility,and Tissue Histology Core supported by the SKCCC CCSG(P30 CA006973)for their services and assistance and the Mass Spectrometry and Proteomics Core facility of the Johns Hopkins School of Medicine.
 Asian Journal of Urology2019年1期
Asian Journal of Urology2019年1期
- Asian Journal of Urology的其它文章
- Application of fluorescence in situ hybridization in the detection of bladder transitional-cell carcinoma:A multi-center clinical study based on Chinese population☆
- Detection of androgen receptor(AR)and AR-V7 in small cell prostate carcinoma:Diagnostic and therapeutic implications
- Prostate tumor neuroendocrine differentiation via EMT:The road less traveled
- Regulatory signaling network in the tumor microenvironment of prostate cancer bone and visceral organ metastases and the development of novel therapeutics
- Combination therapy with androgen deprivation for hormone sensitive prostate cancer:A new frontier
- Potential impact of combined inhibition of 3α-oxidoreductases and 5α-reductases on prostate cancer