
Periodic DFT Study on High Pressure Behavior of Nitrogen-rich Energetic Crystal 4-Amino-3,7-Dinitrotriazolo-[5,1,c][1,2,4] Triazine
2019-11-11 07:59YANGDongfangLIHuiliLIUJinjianZHAOGuozhengLUMing
火炸藥學(xué)報(bào) 2019年5期
YANG Dong-fang, LI Hui-li, LIU Jin-jian, ZHAO Guo-zheng, LU Ming
(1. School of Chemical and Material Science, Shanxi Normal University, Linfen Shanxi 041004, China; 2. School of Chemical Engineering, Nanjing University of Science and Technology, Nanjing 210094, China)
Abstract:The high pressure behavior of nitrogen-rich energetic crystal 4-amino-3,7-dinitrotriazolo-[5,1,c][1,2,4]triazine (DPX-26) in the hydrostatic pressure range of 0—130 GPa was investigated by employing the GGA/PBE-G06 method of periodic density functional theory (DFT). The changes of crystal structure, molecular structure and electronic structure of DPX-26 with pressure were analyzed by calculating lattice parameters (a, b, c), bond lengths, band gaps (ΔEg) and density of states (DOS). The results show that DPX-26 undergoes three significant structure transitions at 81, 82 and 92GPa. The structural transformation occurs at 81GPa with the destruction of intermolecular six membered rings. At 82GPa, the triazole ring is severely distorted and dissociated with the elongation of C4—N7 bond. As the pressure reaches 92 GPa, the bond length of C4—N7 decreases from 0.2324nm to 0.1333nm, and the triazole ring is formed again. The increase of external pressure leads to the delocalization enhencement of the system.
Keywords:quantum chemistry; nitrogen-rich energetic crystal; DFT; high pressure behavior; DPX-26
Introduction
Energetic compounds have a good development prospect and are widely used in both national military and civilian applications. In recent decades, designing new high energetic density compounds, particularly nitrogen-rich energetic compounds, is a new challenge for researchers[1-2]. The nitrogen-rich energetic compound 4-amino-3,7-dinitrotriazolo-[5,1,c][1,2,4] triazine (DPX-26) has been synthesized by Davin et al[3]. The DPX-26 exhibits comparative detonation performance with 1,3,5-trinitro-1,3,5-triazacyclohexane (RDX) but shows a higher thermal stability and lower sensitivity, owing to the strong hydrogen bonds and nitro-π interactions along with planar conjugated structure. Although the synthesis of DPX-26 has been reported, there are still many fundamental and practical problems to be solved, due to the complicated chemical behavior.
To our knowledge, energetic compounds bear tremendous pressure during the detonation process and would undergo phase transitions, structure transformations, decomposition or polymerization[4]. Therefore, describing the changes of molecular structures in energetic crystals under high compressions can contribute to understand more about the detonation mechanism and performance of explosives. Actually, it is scarcely possible to investigate the effect of high pressure on explosives by experimental measurements. However, the theoretical study provides an excellent approach to explore high pressure behavior. And density functional theory (DFT) method has been applied successfully to study the structures and properties of energetic crystals under high hydrostatic pressure[5-8].
In this study, the detailed information about DPX-26 crystal under hydrostatic pressure of 0—130 GPa was obtained by using the periodic DFT calculation. In particular, we focused on the effect of pressure on structural transformations, intermolecular hydrogen interactions and electronic structure.
1 Experimental
Density functional theory (DFT) based on CASTEP code[9]was applied with Vanderbilt-type ultrasoft pseudo-potentials[10]and a plane-wave expansion of the wave functions. The band-by-band conjugate gradient technique was used to minimize total energy of the system with respect to the plane-wave coefficients. The structure was relaxed by the BFGS method. The generalized gradient approximation (GGA)[11-13]was employed to calculate the DPX-26 crystal at 0 K. The cutoff energy of plane wave was set to 600 eV with ak-point grid of 2×2×3. In the process of structural optimization, the total energy was converged to less than 10-5eV, the residual force lower than 0.3 eV/nm, the residual bulk stress under 0.05GPa, and the displacement of atoms below 0.001?. The experimental DPX-26 crystal (CCDC 1481755)[3], used as initial structure, belongs to the monoclinic space groupCcwitha=0.6205nm,b=2.2370nm andc=1.7547nm and contains twelve molecules per unit cell as shown in Fig. 1.
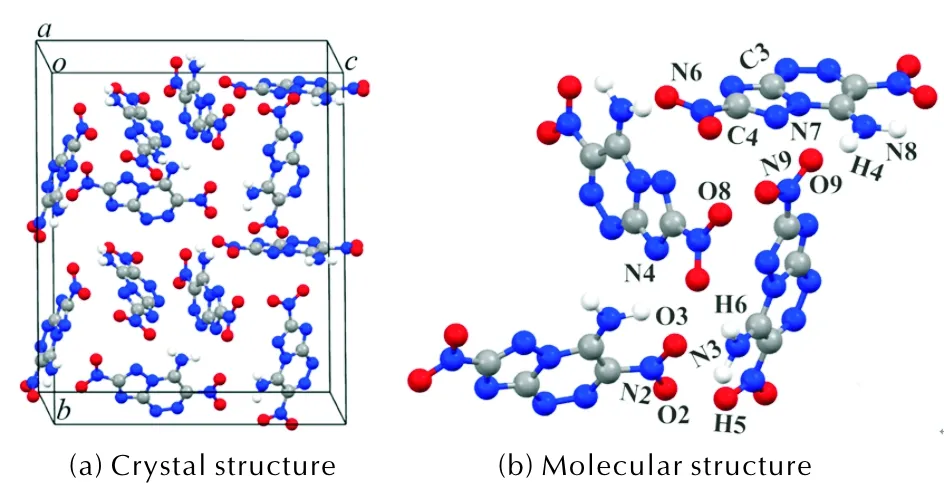
Fig. 1 Crystal structure and atomic number of DPX-26
2 Results and discussion
The generalized gradient approximation (GGA) functional with or without the van der Waals correction were used to relax the DPX-26 crystal totally without constraint at ambient pressure. Table 1 lists the optimized cell parameters with different methods, along with the experimental data for DPX-26. The relative errors of the calculated values to experimental ones show that the lattice constants calculated by GGA/PBE-G06 method agree well with the experimental data, which is lower than other methods. Therefore, GGA/PBE-G06 method was applied to discuss the effect of high pressure in this study.
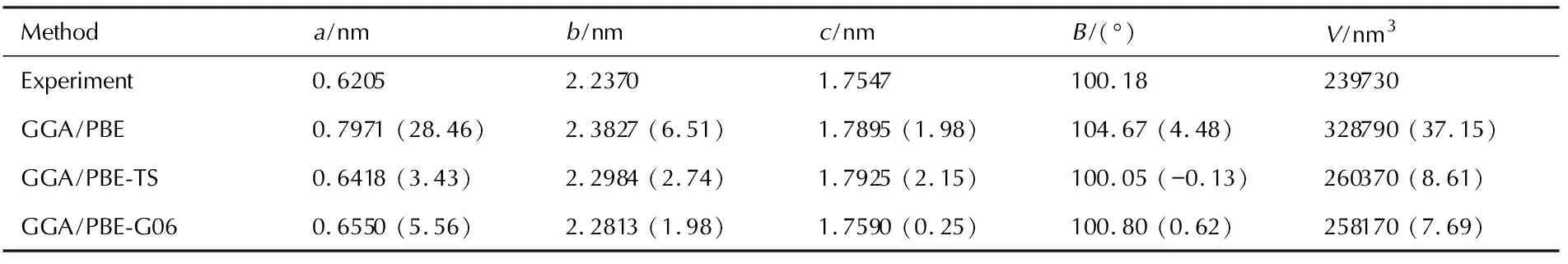
Table 1 Comparison of the relaxed lattice parameters of DPX-26 and the experimental data at ambient conditions
Note: The values in parentheses correspond to the percentage differences relative to the experimental data
2.1 Crystal structure
Fig. 2(a) shows the variation tendency of lattice constantsa,bandcthroughout the whole pressure range from 0 to 130GPa. On the whole,a,bandcdecrease with the augment of pressure, due to the fact that the external pressure is large enough to overcome the intermolecular repulsion along crystallographic directions and makes the crystal structure shrink. And it is clearly found that the curveais flat comparatively. In the low pressure range of 0—80GPa, lattice parameters decrease monotonously with increasing pressure. However, some obvious fluctuations occur under the hydrostatic pressure of 80—130GPa. From 80 to 81 GPa,aincreases from 0.4484 to 0.4595nm anomalously andbdecreases from 1.8372 to 1.7041nm, suggesting that a larger compression occurs in thebdirection at 81GPa. However, at 82GPa, lattice parameters decrease exceptcincreases by 0.0678nm. When the pressure increases to 92GPa,bincreases by 0.0853nm suddenly. All phenomena indicates that large changes in the DPX-26 crystalline have occurred at 81, 82 and 92GPa.
Fig. 2(b) shows the compressibility ratios ofa,bandcunder different pressures. Under low pressures, the intermolecular distance is far and the intermolecular repulsion is weak. Therefore, the crystal is more compressible than that at high pressures. When the pressure increases, the repulsive force becomes strongerso that DPX-26 is difficult to compress. It is apparent from Fig. 2(b) that the compression ratios of lattice constants are different, which confirms that the compressibility alonga-axis,b-axis andc-axis is anisotropic. The sequence of the compressibility along three different directions isa>b>c, except the compression ratio ofbis a little larger thanaduring 85 and 90GPa, suggesting that the cyrstal alongcdirection is much stiffer than the other two directions.
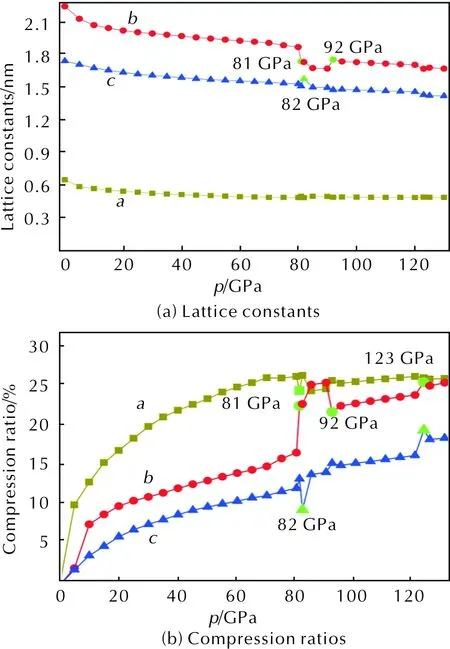
Fig. 2 Lattice constants and compression ratios as functions of pressure
2.2 Molecular structure
The effect of hydrostatic pressure on bond lengths plays an important role in the study of high-pressure behavior. The reason is that external pressure causes obvious transformations in molecular structures and further leads to the changes in chemical property. Fig.3 is the variation of bond lengths with the increasing pressure.
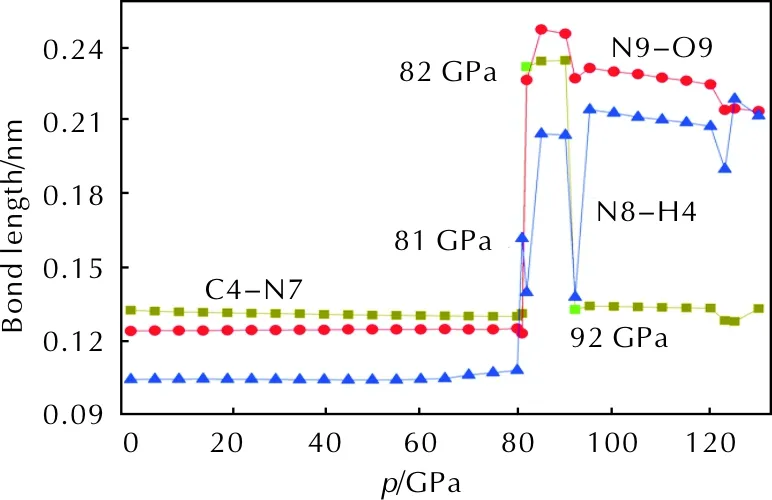
Fig. 3 Variation of the bond lengths with the increasing pressure
It is clear that below 80GPa, the curve is almost horizontal, indicating that the applied compression mostly squeezes out the intermolecular space and causes only a little change in intramolecular geometry. However, when the pressure increases from 80 to 81GPa, the bond length of N8—H4 increases from 0.1084 to 0.1623nm and the torsion angle of H3—C5—C6—H4 changes from 33.6° to 64.7°, implying that the NH2group rotates obviously on the triazine ring. At 82GPa, there is a remarkable change in molecular geometry. The bond length of C4—N7 in the triazole ring elongates to 0.2324nm suddenly. The bond length of N9—O9 increases from 0.1235 to 0.2270nm compared with that at 81GPa, that is to say, the N9—O9 bond breaks above 82GPa. At the same time, the C4—O8 and C3—N9 covalent bonds form, which means the new five-membered ring C4—O8—N9—C3—N6 is formed. All changes attribute to that the molecular structures transform towards more stable configurations under external compression. From 82 to 92GPa, the C4—N7 bond length shortens from 0.2324 to 0.1333nm, which means that the triazole ring rebuilds. It is worth noting that, within the pressure range 80—130GPa, the structural transformations are caused by the increasing of the repulsion between two bonding atoms under compression. This shows that the positions of molecules have changed under external pressure.
The geometrical criteria for the formation of hydrogen bond is that the bond length is in the range of 0.15—0.22nm[14]. Intermolecular hydrogen bonds which consist of the amino groups of one molecule and nitro groups from the other molecules influence the arrangement of molecules in crystal. Therefore, the intermolecular hydrogen bonds are of great importance in DPX-26 crystal. Fig.4 shows the effect of pressure on hydrogen bond lengths.
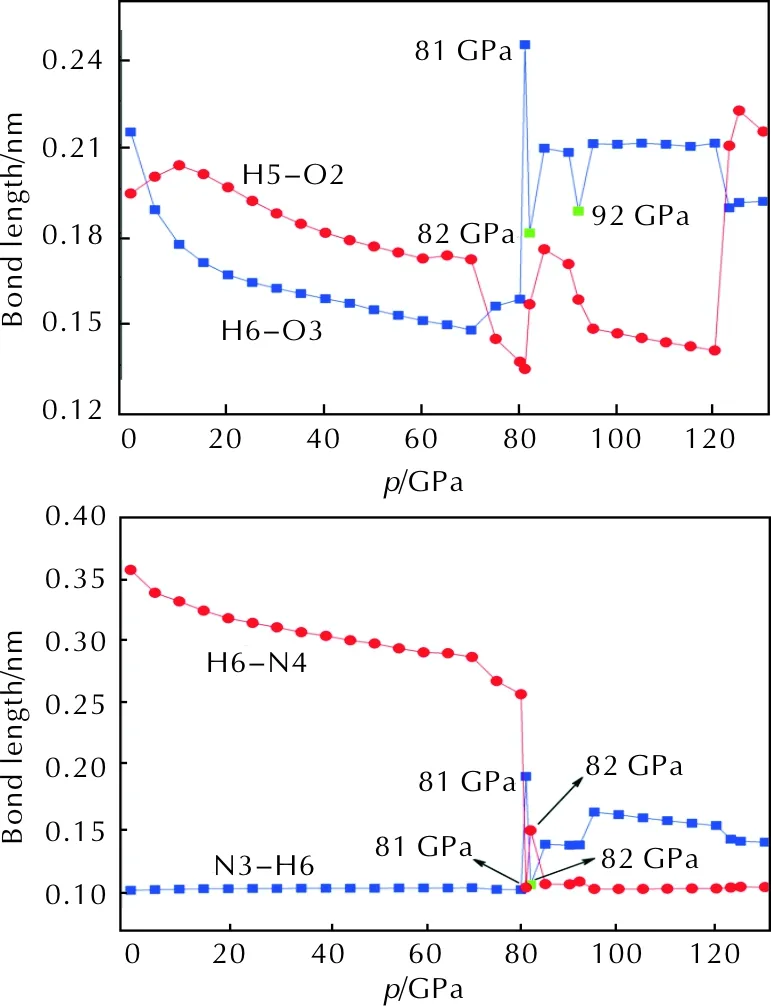
Fig. 4 Variations of bond lengths for intermolecular H-bonds with the increasing pressure
Based on the analysis of Fig. 4, the intermolecular six membered ring N2—O2—H5—N3—H6—O3 forms below 80GPa because of the existence of hydrogen bonds O3…H6 and O2…H5. At 81 GPa, the bond length of O3…H6 reaches 0.2440nm, meaning that the intermolecular six-membered ring is destroyed under the compression. However, the new covalent bond forms between O2 and H5 and the bond length is 0.1337nm. In addition, the distance between N4 and H6 decreases from 0.2582nm to 0.1056nm and the N3—H6 bond length suddenly increases from 0.1035 to 0.1934nm, that is, the hydrogen transfer occurs between the two molecules (H6 transfers from N3 of the NH2group to N4 of the triazole ring). From 81 to 82GPa, the O2—H5 bond length increases by 0.0220nm and O2—H5 bond turns into hydrogen bond. The length of O3…H6 bond decreases to 0.1799nm, therefore, the intermolecular six-membered ring forms once again with a chair conformation. As the same time, the N4—H6 bond elongates to 0.1507nm and turns into hydrogen bond. When the pressure exceeds 92GPa, the length of N4—H6 bond is below 0.1200nm, thus the strong covalent interaction exists between N4 and H6 above 92GPa. The changes of distance between two bonding atoms suggest that the position of molecules has changed, which is in agreement with the result observed from the bond lengths.
In conclusion, the high pressure has a huge influence on the molecular structure in DPX-26 crystal. By analyzing the bond lengths, it is easy to find that the molecules undergo structure changes under external pressure, especially at 81, 82 and 92GPa. Fig.5 summarizes the transformations of molecules under different pressures.

Fig. 5 The variations of molecular structures under different pressures. Gray, blue, red and white spheres stand for carbon, nitrogen, oxygen and hydrogen atoms, respectively
2.3 Electronic structure
Studying the electronic structure of a solid material facilitates the deeper investigation of the crystalline structure and physical properties. The energy gap (ΔEg) is a significant parameter to characterize the electronic structure of energetic crystal. Fig. 6 exhibits the changes of ΔEgwith the increasing pressure.
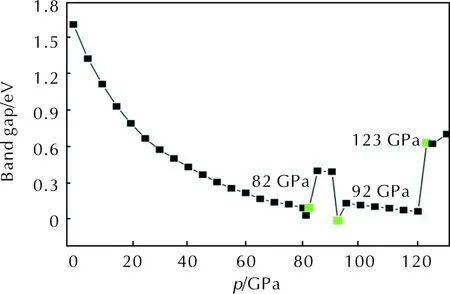
Fig. 6 Band gap of DPX-26 crystal as a function of pressure
From Fig. 6, when the pressure is lower than 81GPa, ΔEgdecreases successively from 1.615 to 0.042eV, due to the reduction in intermolecular distance. It is suggested that electrons are easy to transfer from occupied valence bands to empty conduction bands under external pressure. The reason is that the narrower the intermolecular space, the more electron overlap of energy bands. Additionally, the DPX-26 undergoes an electronic phase transition from a semiconductor to a metallic system. In the range of 82—90GPa, by the reason of deformation of triazole ring and formation of intermolecular six-membered ring, ΔEgincreases dramatically, which leads to the energy bands shift towards the higher energy region. At 92GPa, ΔEgdecreases to 0eV completely, owing to the structure transformation, that is, the triazole ring builds once more.
The density of states (DOS) is an effective tool to understand the bond nature and electronic structure under different pressures. Fig. 7 depicts the DOS for crystalline DPX-26 at different pressures.

Fig. 7 The DOS for crystalline DPX-26 at different pressures, s, p and total states are shown as dot, dash and solid curves, respectively
The curves of DOS behave as obvious peaks in the pressure range of 0 80GPa. As the pressure increases, the peaks become low and wide, which explains that the electronic delocalization increases gradually in bulk DPX-26. Thus, the DPX-26 becomes a conductor under high compression. It is worth while to note that thepstates play an essential role in the sharp peaks of valence and conduction band near the Fermi level, which signifies thatpstates have a significant effect on the chemical reaction of DPX-26 crystal. Besides, the conduction bands tend to shift towards lower energy region with the increasing pressure, which leads to the band gap reduce. The DOS peaks, at 92GPa, connect together near the Fermi energy level because the band gap reduces to 0eV, suggesting that the electrons can move freely from the valence band to conduction band.
3 Conclusion
DFT periodic calculations have been applied to explore the effect of external pressure on DPX-26 crystal in the range of 0—130GPa. And the GGA/PBE-G06 functional was adopted to perform the computation. The conclusion is drawn as follows:
(1) The crystalline DPX-26 undergoes three distinct molecular structure transformations at 81, 82 and 92GPa. At 81GPa, the planarity of molecules is destroyed, meanwhile, the intermolecular six-membered ring ruptures. When 82GPa is applied, the length of C4—N7 bond is more than 0.22nm, and the triazole ring cleavages. The bond length of N4—H6 increases to 0.1507nm, and the covalent interaction translates into hydrogen bonding. At 92GPa, the triazole ring rebuilds because C4—N7 bond length shortens from 0.2324 to 0.1333nm.
(2) The external pressure leads to the increase of electronic delocalization in bulk DPX-26. All analysis shows that the pressure-induced transformations in the molecular structure and changes in electronic structure occur at 81, 82 and 92GPa.
