
Cathepsins in neuronal plasticity
2021-08-20 12:36AmandaPhuongTranJerrySilver
中國神經(jīng)再生研究(英文版) 2021年1期
Amanda Phuong Tran, Jerry Silver
Abstract Proteases comprise a variety of enzymes defined by their ability to catalytically hydrolyze the peptide bonds of other proteins, resulting in protein lysis. Cathepsins, specifically,encompass a class of at least twenty proteases with potent endopeptidase activity. They are located subcellularly in lysosomes, organelles responsible for the cell’s degradative and autophagic processes, and are vital for normal lysosomal function. Although cathepsins are involved in a multitude of cell signaling activities, this chapter will focus on the role of cathepsins (with a special emphasis on Cathepsin B) in neuronal plasticity. We will broadly define what is known about regulation of cathepsins in the central nervous system and compare this with their dysregulation after injury or disease. Importantly, we will delineate what is currently known about the role of cathepsins in axon regeneration and plasticity after spinal cord injury. It is well established that normal cathepsin activity is integral to the function of lysosomes. Without normal lysosomal function, autophagy and other homeostatic cellular processes become dysregulated resulting in axon dystrophy.Furthermore, controlled activation of cathepsins at specialized neuronal structures such as axonal growth cones and dendritic spines have been positively implicated in their plasticity.This chapter will end with a perspective on the consequences of cathepsin dysregulation versus controlled, localized regulation to clarify how cathepsins can contribute to both neuronal plasticity and neurodegeneration.
Key Words: axon regeneration; cathepsin; CSPGs; extracellular matrix; growth cone;lysosomes; neuronal plasticity; protease; remodeling; spinal cord injury; synaptogenesis
Introduction
Named by Willst?tter and Bamann (1929) for theirkathepseinor “digestive” properties, cathepsins were first noted for their proteolytic abilities when they were initially derived from leukocytes (Verma et al., 2016). Since their discovery in 1929, we have come to understand that cathepsins can be further divided by their catalytic mechanisms and preferred substrates. These subtypes, totaling more than 20, include serine, cysteine, and aspartyl endopeptidases. At least some subtypes of cathepsins are ubiquitously expressed in lysosomes, a membrane-enclosed organelle responsible for digesting proteins and other biological polymers, or endosomes of all cell types (Turk et al., 2012; Cocchiaro et al.,2017). The subcellular location of cathepsins is particularly notable since they require low pH environments for activation(Jerala et al., 1998; Turk et al., 2012; Colacurcio and Nixon,2016). Lysosomes are able to sequester potently degradative cathepsins as membrane bound V-type ATP-powered H+pumps which helps maintain these subcellular structures at a lower pH than the cytoplasm (Kubisch et al., 2014; Colacurcio and Nixon, 2016). Cathepsins are integral to various processes associated with lysosomes including protein degradation,metabolism, turnover, antigen presentation, cell death signaling, phagocytosis, growth factor receptor recycling and,notably, autophagy (Turk et al., 2012). As such, dysregulated cathepsin expression or activity is responsible for a range of autoimmune, inflammatory (Patel et al., 2018), and even neurodegenerative disorders (Stoka et al., 2016).
In this review, we will first begin with a broad introduction into the regulation, activity, and physiological functions of cathepsins. We then describe how cathepsins, notably cathepsin B, becomes dysregulated following spinal cord injury. Dysregulation of cathepsins in the context of spinal cord injury not only expands inflammatory processes and furtherstissue damage, but also perturbs homeostatic processes such as autophagy and lysosomal function. Cathepsin dysregulation has been implicated in neurodegenerative disorders such as Alzheimer’s disease. However, under normal conditions,cathepsins are vital for the role they play in neuronal plasticity.Specifically, cathepsins B and D have been noted for their roles in extracellular matrix remodeling. In the central nervous system (CNS), this phenomenon is particularly interesting in regulating neuronal plasticity including axon regeneration and dendritic spine growth. We will review how controlled cathepsin B secretion by the axon growth cone is necessary for extracellular matrix remodeling and degradation of regeneration-inhibitory chondroitin sulfate proteoglycans(CSPGs) after spinal cord injury. We will then describe recent findings delineating the role of cathepsin B in activitydependent dendritic spine remodeling. Understanding the role of cathepsin B in axon outgrowth and dendritic spine dynamics will help to elucidate how neurons endogenously enhance neuronal plasticity. Perhaps these mechanisms can one day be exploited to the benefit of enhancing neuronal plasticity following spinal cord injury or neurodegenerative disease.
A search of PubMed between October and December 2019 for axon regeneration, axon growth, axon sprouting, cathepsins,synaptogenesis, structural plasticity, neural plasticity, spinal cord injury was conducted.
Regulation, Activity, and Physiological Functions of Cathepsins
All protease activity must be tightly regulated to prevent detrimental off-target effects that may perturb cell signaling,damage cell membrane integrity or normal protein function,and even perpetuate cell death signaling (Khalkhali-Ellis and Hendrix, 2014). Regulation of cathepsins occurs in stages starting with transcriptional activation and translation followed by precise subcellular localization and then zymogen activation and de-activation (Figure 1) (Barrett, 2002; Bai and Pfaff, 2011). However, it is important to think of these regulatory stages existing as a network instead of a simplified sequential one-step-after-another cascade. Thus, more than one cathepsin is responsible for activating another and each cathepsin often interacts with more than one substrate including other proteases. Regarding protease regulation in general, it is essential to consider the timing and specific localization of protease activity in addition to the specific upstream activators. Moreover, the context of the cellular environment must be considered in these regulatory events.Even tight regulation at each step of cathepsin production can be skewed toward dysregulation in a context of inflammation.
To begin, cathepsin production at the level of transcriptional activation is a convenient point where its regulation can be enforced. Increased expression of cathepsin B in cancer progression, for example, includes use of alternative promoters, increased transcription and alternative splicing,and increased stability and translation of cathepsin B mRNA(Podgorski and Sloane, 2003). The promoter region of cathepsin B includes the proto-oncogenes, Sp1 and Ets1 protein binding sites and, as such, these proteins can regulate transcription effectively (Yan et al., 2000; Aggarwal and Sloane, 2014). Signaling pathways and their downstream transcription factors responsible for upregulating cathepsin B/D transcripts, especially in the context of cancer, include the lysosomal upregulating transcription factor EB (Sardiello et al., 2009; Settembre et al., 2011), and pathways related to inflammation signaling such as NF-kappa B (Hamer et al., 2012) and beta-catenin (Herencia et al., 2012). Of note,upregulation of transcripts as assessed through qPCR,microarray, or next generation sequencing alone may not fully reveal biological significance especially in the context ofin vitroinjury models and should be further validated.Cathepsins are then translated and processed by the endoplasmic reticulum in catalytically inactive pro-forms of the protease called zymogens. Post-translational changes within the Golgi apparatus then occur and include addition of sugar linkages, specifically N-glycosidic-linked oligosaacaride chains with mannose 6-phosphate residues which are critical for translocation of the cathepsins to the lysosome (Braulke and Bonifacino, 2009) and keeping the cathepsin in its inactive form during transport (Hasilik et al., 2009). In addition to glycosylation, other potentially reversible post-translational modifications including acetylation, hydrocarbonylation,ubiquitylation, and so on occur to modify cathepsin transport,longevity, or function (Katunuma, 2010). Following posttranslational modifications, cathepsins are packaged into endosomes before they are delivered to the lysosome (Turk et al., 2012) where their intended substrates are found (Barrett,2002). For the cathepsin zymogen to be fully activated, it must be present in the acidic environment of the lysosome, which is maintained by membrane-bound V-type H+ion pumps (Hasilik et al., 1982). These pumps hold the subcellular environment at a pH of 5 compared to the cytoplasmic pH of 7.2 (Aggarwal and Sloane, 2014). Within the lysosome, cathepsin zymogens are cleaved at the pro-domains (Olson and Joyce, 2015). This occurs either through auto-activation signaled by the acidic environment to shed the N-terminal propeptide domain or by protease-induced activation. Auto-activation is seen in cathepsins B, D, L, X, and Y among others. Activation through cleavage of the propeptide domain can also be performed by other activated cathepsins (Laurent-Matha et al., 2006; Olson and Joyce, 2015). Some negatively-charged molecules are also involved in the processing of cathepsins in physiological environments. For example, the auto-activation of cathepsin B is accelerated by sulfated glycosaminoglycans, the highly negatively charged sugar moieties found on axon-inhibitory CSPGs (Caglic et al., 2007).
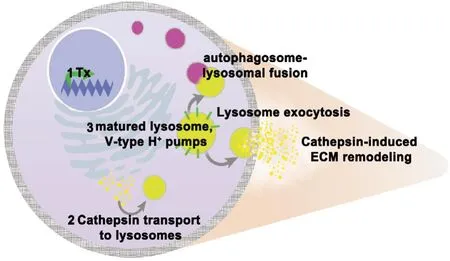
Figure 1 |Regulation, activity, and physiological functions of cathepsins.Tight regulation of cathepsins (yellow) occurs in various stages of its production including 1) Transcriptional activation, 2) production as zymogens that are activated in 3) the acidified environment of the mature lysosome.Lysosomes (depicted as yellow organelles) possess V-Type H+ ion pumps(green) that hold the subcellular environment around pH 5. Within the lysosome, zymogen cathepsins are activated once the pro-domains are cleaved. Matured cathepsins play a significant role in cellular homeostasis including maintaining autophagic flux through fusion with autophagosomes(pink) to degrade and recycle proteins. Lysosomes may also be exocytosed where released cathepsins activate other proteases or themselves to remodel the extracellular matrix (ECM). Tx: Transcription.
While acidity is important for cathepsin activation, cathepsin B was demonstrated to perform some catalytic activity at neutral pH as well, such as in the extracellular space (Mort et al.,1984; Buck et al., 1992; Linebaugh et al., 1999). Furthermore,cathepsin B was observed in other cellular structures beyond the lysosome such as secretory vesicle-enriched fractions(Sendler et al., 2016). This suggests that cathepsins may be excreted through the secretory autophagic signaling pathway,which was documented to enable assortment and secretion of lysosomal contents into the extracellular space (Sasaki and Yoshida, 2015). Moreover, cathepsin B localizes in membrane microdomains including the epithelial lining (Jokimaa, 2001),and caveolae in both tumor and endothelial cells (Cavallo-Medved et al., 2003; 2009). This is notable because caveolae,or little caves, of the cell are small vesicular invaginations of the cell membrane where signaling molecules gather to assemble signaling complexes (Schlegel et al., 1998). Here,they are also well positioned to degrade the local extracellular matrix. Exocytosis of cathepsin B was observed in other cell types such as fibroblasts or endothelial cells in an annexin II-regulated manner (Mohamed and Sloane, 2006) and by neurons as well (Padamsey et al., 2017; Tran et al., 2018a).Thus, while the majority of cathepsins maintain activity in lysosomes, some subtypes of cathepsins can be found in endosomes concentrated in specialized cellular niches.Cathepsins K, W, and S show specifically restricted cellular distributions in either osteoclasts, epithelial cells, or synovial fibroblasts respectively (Hou et al., 2001; Turk et al., 2012).Translocation and localization of cathepsins in subcellular niches therefore constitute an effective way to enable their controlled regulation.
Within the family of cathepsins there are cysteine, serine,and aspartyl proteases. Cysteine proteases include cathepsins B, C, F, H, K, L, O, S, V, W, and Z (also called X). As the name suggests, these cysteine proteases cleave peptide bonds in which the cysteine residue of the substrate serves as the nucleophilic amino acid at the protease’s active site. The cathepsin family also includes serine (A, G) and aspartyl (D,E) proteases. Cysteine cathepsins have been noted for their promiscuous substrate selectivity based on their specific endopeptidase activity on common amino acids (Biniossek et al., 2011). For example, cathepsin L has the potential to cleave proteins at 845 potential sites in the most comprehensive proteomic protease cleavage site list to date (Biniossek et al.,2011). Included in this range of substrates are most certainly inhibitory components of the extracellular matrix, enabling axon terminal remodeling and plasticity in the CNS. Most cathepsins target extracellular matrix proteins for degradation(Olson and Joyce, 2015). Cathepsin B, for example, degrades collagen type IV, laminin and fibronectin (Buck et al., 1992).Cathepsins K, L, S, F, and V also show varying potencies of elastin, collagen, and proteoglycan catalysis (Turk et al., 2012).Of particular interest to the CNS extracellular matrix after injury, cathepsin B cleaves axon growth -inhibitory aggrecan(Fosang et al., 1992, 1995; Mort et al., 1998). Cathepsins can degrade a wide range of substrates, activate other proteases through cleavage of their pro-peptide domains,and are able to auto- activate other cathepsins thus existing in an interesting “hub-like” status within the cell’s proteolytic network. Cathepsin B, for example, is able to activate a variety of other proteases such as matrix metalloprotease (MMP)-2, 3, 9, uPA, and even other cathepsins (Mason and Joyce,2011). Thus, cathepsin B represents a highly integrated node in which a proteolytic cascade leads to remodeling of the extracellular matrix. However, because of this hub status and substrate promiscuity, proteases like cathepsin B must betightly regulated to prevent potentially injurious uncontrolled and non-specific degradative activity.
In addition to the subcellular localization strategy of regulation described above, additional controls can be accomplished through translation of endogenous cathepsin inhibitors such as cystatins, serpins, and thyropins, which competitively bind to a specific cathepsin catalytic domains to neutralize activity in a reversible fashion (Aggarwal and Sloane, 2014). Based on the kinetics of these endogenous inhibitors and their specificity to particular cathepsins, they can be categorized as either emergency, buffering, or threshold-type inhibitors(Turk et al., 2012). Stefins (part of the cystatin type I family)are found throughout the cell’s cytoplasm (Chung et al., 2016)as an emergency measure against lyosomal leakiness (Ma et al., 2017) or dysregulation to prevent damaging cathepsin activity. Stefin A, B (also called cystatin B), and cystatins D and F inhibit cathepsin B (Ochieng and Chaudhuri, 2010).The dysregulation of cystatins is also implicated in human diseases as they perturb regulation of cathepsins. Constitutive knockout of cystatin B is one such example, but although genetic knockout of cystatin b was implicated in Unverricht-Lundborg type progressive myoclonus epilepsy (Eldridge et al., 1983; Joensuu et al., 2014), little is known about the roles cystatin b may play in CNS homeostasis or how it is regulated.However, a few studies suggest that like the cathepsins, its expression is also temporally and spatially regulated to effect extracellular matrix remodeling in development. For example,in the developing rat cerebellum, cystatin B expression is undetectable in radial glial cells as visualized through brain lipid binding protein immunostaining at seven days. By 90 days, brain lipid binding protein and cystatin B immunostaining colocalizes along the glial somata and developing axonal projections (Riccio et al., 2005). This may correlate with an increase in activity of cathepsin B and other proteases, such as those from the MMPs (Small and Crawford, 2016). Such proteases may function during development to remodel the extracellular matrix and allow for axon migration while preventing potentially damaging cathepsin B activity in the cytoplasm.
Once activated, cathepsins are responsible for a variety of functions to maintain homeostasis including protein degradation and turnover. In conjunction with the ubiquitin proteasome system, the lysosomal system allows for degradation of selected or damaged proteins. Lysosomes are fed proteins destined for degradation in two ways:endocytosis from the extracellular matrix or through autophagosome fusion at the end stage of autophagy.Endosomes are formed through endocytosis of the cellular membrane where signaling or extracellular components such as integrins (Rainero, 2018), gelatin and collagen (Porter et al., 2013) among others are diverted into lysosomes for degradation. This process aids in extracellular matrix remodeling. Protein degradation is especially important in the context of neurodegenerative disorders. Loss of this homeostatic mechanism results in protein accumulation that disproportionately affects post-mitotic neurons and leads to the development of neurodegenerative disorders such as frontotemporal dementia, Alzheimer’s disease, Huntington’s disease and others. Drugs that specifically enhance the lysosomal degradation pathway have been proposed as an effective, broad strategy against protein accumulation and certain neurodegenerative disorders (Bahr, 2009). Beyond protein degradation and turnover, cathepsins have been noted for a myriad of other functions including antigen presentation,adaptive immunity, peptide activation for cell signaling,cellular differentiation, apoptosis, and beyond (Olson and Joyce, 2015). As such, dysregulation of cathepsins specifically is implicated in a myriad of diseases from tumorigenesis and cancer cell metastasis to neurodegeneration such as in Alzhemier’s disease (Butler et al., 2011), Huntington’s disease (Liang et al., 2011), and Parkinson’s disease (Qiao et al., 2008). In the proceeding sections, we will review how dysregulation of cathepsins occurs following spinal cord injury.We will further explicate the role of cathepsins, especially in extracellular matrix remodeling, and how these proteases affect axonal and synaptic plasticity.
Cathepsin Dysregulation after Traumatic Spinal Cord Injury
In contrast to the normally tightly controlled temporalspatial regulation of proteases, promiscuous and often rampant protease activation has been described in several models of CNS injury and neurodegeneration (Figure 2). In this section, we will describe the sequelae of events resulting in dysregulation of cathepsins following traumatic spinal cord injury. Most of the molecular events resulting from inflammation, however, can be broadly applied to other CNS injuries or degenerative events where inflammation also occurs. Although rampant protease activity and dysregulation of cathepsins, and more broadly, protease dysregulation incurs tissue damage and cell death acutely, we must stress that this is a sequelae of tissue damage occurring in a diseased or injury state in contrast to highly regulated levels of cathepsin activity during homeostasis.
To begin, traumatic injuries to the spinal cord immediately inflict damage to cellular integrity at the lesion epicenter.Specifically, this includes lysosomal rupture or membrane permeabilization (Figure 2) (Yamashima and Oikawa, 2009;Lipton, 2013). Cathepsins are released from damaged lysosomes into the cytoplasm where they initiate apoptotic cascades and additionally leak into the extracellular matrix where they further damage surrounding cells and tissues and play an executory role in apoptoic and necrotic cell death (Tang et al., 2019). In the ensuing release and massive upregulation of calcium after spinal cord injury, cathepsins are further activated and contribute to injury (Ray et al., 2011;Orem et al., 2017). Upregulated calcium was hypothesized to contribute to neuronal death following ischemic injury, which is observed in traumatic spinal cord injury after rupturing of blood vessels and resulting in hemorrhage. In the calpaincathepsin hypothesis, an increase of intracellular calcium resulting from oxidative injury causes downstream lysosomal rupture, opening the floodgates to cytoplasmic release of activated cathepsins and subsequent neuronal death(Yamashima, 2000, 2004, 2013). Furthermore, oxidative stress was observed to translocate lysosomal cathepsin D from axons to the lysosomes where they accumulate and further potentiate apoptosis (Miura et al., 2010).
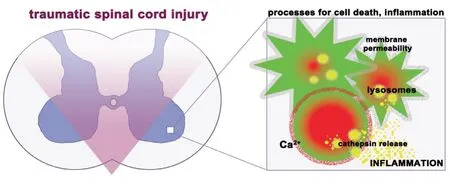
Figure 2 |Cathepsin dysregulation after traumatic spinal cord injury.Following traumatic injury to the spinal cord, rampant protease activation results in tissue damage, cell death, and inflammation. Upregulated intracellular calcium incurred from oxidative injury causes lysosomal rupture and release of activated cathepsins. Released cathepsins induce membrane permeability, cell death, and release and activation of proteases in the extracellular space. Cell death releases alarmins, a molecular signal that initiates the inflammatory cascade. This perpetuates a positive feedback loop that includes a protease storm where dysregulated protease activity furtherstissue damage.
Not only does the initial necrosis of cells at the lesion epicenter release cathepsins, but alarmins are introduced into the extracellular matrix as well (Gadani et al., 2015a,b). Alarmins initiate the cascade of inflammatory signaling radiating from the lesion epicenter and penumbra, which causes a positive feedback mechanism that releases more proteases and further cell death and release of even more alarmins. Thus, the inflammatory processes incurred by traumatic spinal cord injury initiates a “protease storm” where promiscuous and non-regulated protease activity is unleashed to instigate tissue damage and further the inflammatory cascade (Noble et al., 2002; Veeravalli et al., 2012; Brkic et al.,2015). As proteases are ubiquitously expressed, upregulated protease transcription and secretion was noted in a variety of different cell types following injury including immune cells, oligodendrocyte progenitor cells, astrocytes, and even neurons (Brkic et al., 2015). For example, a wide range of pro-apoptotic proteases released following impact and subsequent cellular damage can exacerbate neuronal and glial death around the lesion epicenter (Veeravalli et al., 2012).These include upregulated mRNA and protein levels of MMPs-3, 7, 9, 10, 11, 13, 19, and 20 and other proteases which have been reported to become upregulated as quickly as 24 hours after spinal cord injury and persisting for 1 week or more (Wells et al., 2003). The addition of other proteases upregulated following injury is noteworthy because they work in a network with cathepsins to activate each other. This ultimately helps to spread inflammatory signaling, tissue and cellular damage, and death after the initial traumatic injury event.The spread of inflammation, however, becomes quenched by the development of the fibroblastic/glial scar composed of reactive astrocytes, NG2+progenitor oligodendrocytes,pericytes/meningeal or endothelial fibroblasts, and other glia(Tran et al., 2018b). While formation of the fibrotic glial scar helps to stabilize the lesion epicenter and prevent the further spread of inflammation throughout the cord, it ultimately remodels the cellular architecture and extracellular matrix of the post-injury environment in such a way as to chronically inhibit axon regeneration and return of function. Primarily,at the molecular level, this is accomplished through the upregulation by reactive astrocytes and other glia of a family of axon growth inhibitory CSPGs, (Zhang et al., 2006; Buss et al., 2009; Cregg et al., 2014). Exactly how cathepsin release by neurons can lead to degradation and dis-inhibition of CSPGs will be further discussed in the section below. Of note, there are other molecular strategies by which glia and neurons reduce protease activity. These include 1) upregulated expression of tissue inhibitors of metalloproteases that inhibit the family of MMPs (Bode et al., 1999); 2) increased presence of cystatins that inhibit cathepsins (Ochieng and Chaudhuri,2010) or 3) post-translational modifications of the proteases themselves (Madzharova et al., 2019).
An upregulation of cathepsins B and D was first observed following spinal cord injury by Banik et al. (1986). They are now known to increase after a variety of different types of neurotrauma (Abou-El-Hassan et al., 2017). In fact, in a neuroproteomics screen, cathepsin D was identified as a major biomarker that predicts the severity of human spinal cord injury (Moghieb et al., 2016). Cathepsin D protein was noted at its highest level at 7 days post-injury in rats after spinal cord injury as detected by western blotting of lesioned spinal cord lysate (Zhang et al., 2014). Most neurons and some glia express cathepsin D in the normal spinal cord; however,following injury cathepsin D is highly upregulated in infiltrating macrophages/resident microglia after rat spinal cord injury(Moon et al., 2008). This has implications in the activation of macrophages including phagocytosis and lysosomal activity after injury. Likewise, cathepsin B mRNA expression is increased following contusive spinal cord injury as early as 2 days post- injury (Ellis et al., 2005). Cathepsin B enzymatic activity also peaks at 5-7 days post-injury with the greatest contribution from immune cells at the lesion epicenter (Ellis et al., 2005). Immune cell expression of increased cathepsins incurs longterm consequences well after injury. Cathepsin S, for example, upregulated by microglia following spinal cord injury contributes to chronic neuropathic pain (Clark et al., 2007a). Likewise, microglial cathepsin B was observed to contribute to the initiation of peripheral inflammationinduced chronic pain (Sun et al., 2012). Cathepsins have also been known to activate metalloproteases, which have been implicated in furthering tissue damage after injury through indiscriminate degradation of the components of the blood spinal cord barrier (Noble et al., 2002; Yong et al., 2007)including proteins of the basal lamina that keeps the blood spinal cord barrier chronically leaky (Veeravalli et al., 2012).
In addition to playing deleterious roles in tissue damage following traumatic spinal cord injury, dysregulated cathepsins have been characterized in other forms of neurotrauma and neurodegeneration where inflammatory processes are also upregulated. For example, cathepsin B activity is increased and the enzyme is released into the extracellular matrix after traumatic brain injury (Hook et al., 2015). In Alzheimer’s disease where chronic inflammation occurs and lysosomal function is also compromised (Cataldo and Nixon, 1990),cathepsin B may also play a role. In Alzheimer’s disease, axons develop well-characterized dystrophic morphology around senile plaques filled with cathepsin-laden lysosomes (Lee et al., 2011). Cathepsin B activity improved β-amylodiosis and learning and memory in models of mouse Alzheimer’s disease (Wang et al., 2012; Embury et al., 2017; Farizatto et al., 2017) while genetic knockout of cathepsin B leads to lysosomal dysfunction and enhanced amyloid-β and amyloid precursor protein load (Cermak et al., 2016). Aside from degrading CSPGs, cathepsin B in neurons further digested amyloid-beta in mouse models of Alzheimer’s disease (Butler et al., 2011) and functionally improved memory (Yang et al.,2011). The ability of cathepsins B and D to reduce mutant huntingtin accumulation and toxicity in neurons has also been noted (Liang et al., 2011). However, cathepsin B activity was implicated as a key driver of inflammation when expressed by microglia (Nakanishi, 2020). Cathepsin B was observed to act like the beta-secretase BACE1 to produce pathological cleaved forms of amyloid precursor protein and pyroglutamate A-beta peptides when it is secreted by neurons (Schechter and Ziv,2011; Hook et al., 2014; Bernstein and Keilhoff, 2018).
The ability of cathepsin B to simultaneously improve function and drive inflammation in Alzheimer’s disease perfectly encapsulates the duality of cathepsin activity in regeneration and degeneration. While dysregulated protease release and activity clearly have potent and detrimental effects in the inflammatory milieu, greater emphasis must be placed on the endogenous actions of proteases that are highly regulated in a temporal-spatial manner to allow for cellular homeostasis. Emphasized in the following sections is the use of proteases well after pro-inflammatory events to degrade inhibitory CSPGs that have been upregulated during CNS insult. A variety of different proteases, for example,have been specifically secreted by axons to enhance CSPGrestricted plasticity in adulthood.
The Role of Cathepsins in Axon Homeostasis and Regeneration
Cathepsins largely affect axon outgrowth in two ways: by promoting axonal homeostasis to maintain a healthy growth motor or by its secretion into the extracellular matrix at the growth cone to degrade axon-inhibitory constituents of the extracellular matrix such as CSPGs (Figure 3). Local secretion of cathepsins to degrade inhibitory CSPGs then, in turn,promotes axon regeneration/sprouting. Some other strategies by which cathepsins modulate neurite outgrowth or promote axon regeneration will also be described. Here, we will refer to axon regeneration as the de novo growth as well as the regrowth of a previously transected axon (Steward et al., 2003;Tuszynski and Steward, 2012). Also notable is the sprouting of axon collaterals from nearby, undamaged axons.
Cathepsins are well-known as lysosomal proteases that aid in proteolytic degradation emanating from within these specialized structures (Olson and Joyce, 2015) and have been noted as pro-regenerative proteins in peripheral axons, which are capable of regeneratingin vitroandin vivo(Kubo et al.,2004; Gordon et al., 2009; H?ke and Brushart, 2010). The distribution of cathepsins in neurons is also unique compared to other cell types as neurons possess specialized structures such as long axons and dendritic spinous processes. Indeed,immunostaining for lysosomes and other acidic vesicles using the acidic pH dye, LysoTracker, revealed colocalizion of the majority of cathepsin B within these structures which are found not only in the somata of peripheral neurons,but throughout their axons and growth cones as well(Gowrishankar et al., 2017; Cheng et al., 2018; Tran et al.,2018a). Farel-Becker et al. (2020) have shown that cathepsins B and D are actively transported to the axon tip where they are especially abundant. Further, lysosomal/endosomal exocytosis mediated by calcium-induced synaptotagmin VII interacting with VAMP7 was found to be necessary for peripheral neurite outgrowth and arborization (Arantes and Andrews, 2006). Work by the Nixon group further highlighted the importance of lysosomal function in axon growth and homeostasis. For instance, inhibition of lysosomal proteolysis through de-acidification or cathepsin inhibition resulted in an accumulation of autophagic vacuoles in bulbous swellings, and ultimately, dystrophic axons (Lee et al., 2011). This was also observed in cathepsin D and cathepsins B and L constitutive knockout mouse lines where accumulation of LC3+(a marker of autophagosomes) vacuolar structures were found along the axons and neurons within the CNS of the mutant (Koike et al.,2005).
Additionally, cathepsin B activity as a part of lysosomal function and autophagy are inextricably linked as reduced or inhibited cathepsin B activity through Ca074-me or overexpressing its inhibitor, cystatin B, was reported to impair autophagy(Lamore and Wondrak, 2011; Tatti et al., 2012; Soori et al.,2016) and enhance lysosomal dysfunction (Mizunoe et al.,2017). At the end stage of autophagy (including chaperonmediated autophagy and microautophagy), lysosomal fusion with mature autophagosomes occurs to degrade the contents of autophagic bodies. Recent studies have come to better appreciate this connection as blockage of autophagosomelysosomal fusion led to diminished lysosomal activation (Zhou et al., 2013) and inhibition of lysosomal activation in the face of sustained autophagy resulted in neuritic dystrophy (Bordi et al., 2016). Simply put, the failure of lysosomal transport and hydrolase activity, as supported by cathepsins, induces autophagic stress (Farfel-Becker et al., 2019, 2020), inhibits axon regeneration, and in extreme cases induces neuronal apoptosis (Ferguson, 2018). The link between autophagosomelysosomal fusion and axonal dystrophy helps to molecularly characterize the heretofore unidentified cycling vesicles first discovered by Tom et al. to be a hallmark of dystrophic, or stalled non-regenerating, growth cones. These dystrophic growth cones were effectively trapped by a gradient of CSPGs and failed to extend further (Tom et al., 2004). Recent findings by Sakamoto et al. (2019) have found that CSPGs inhibit this autophagosome and lysosomal fusion step and that these bubbling vesicles are the result of autophagic vesicles failing to fuse with lysosomes.
Connecting Extracellular Chondroitin Sulfate Proteoglycans with Lysosomes and Autophagy
CSPGs are proteoglycans upregulated in the extracellular matrix after trauma or inflammation to the CNS and are potent inhibitors of axon regeneration or neuronal plasticity after injury. These proteoglycans bind to their cognate receptor, protein tyrosine phosphatase sigma (PTPσ) and in doing so, activate its phosphatase activity (Shen et al., 2009;Coles et al., 2011; Wu et al., 2017). One of the secondary effectors targeted by PTPσ for de-phosphorylation is cortactin,an actin-binding protein, which reduces its ability to stabilize the actin cytoskeletal infrastructure required to enable autophagosome/lysosomal fusion (Sakamoto et al., 2019).In addition to regulating this fusion step, cortactin regulates lysosomal maturation and trafficking, and regulates protease secretion into the extracellular matrix (Clark et al., 2007b;Clark and Weaver, 2008; Kirkbride et al., 2012). Whether cortactin specifically regulates lysosomal-induced secretion of cathepsins into the extracellular matrix itself will require future studies. Work in our lab, additionally, begins to show that cathepsin B is enhanced in the axons of PTPσ null neurons compared to wild type neurons (Figure 3) (Tran et al.,2018a). In all, this highly interesting and newly appreciated link between lysosomal-autophagic flux, CSPGs, and axon inhibition will require further study. Thus far, however, it is evident that CSPGs serve not only as a molecular barrier to axon regeneration, but they also dysregulate normal axon homeostasis (i.e., disrupted autophagic flux and decreased cathepsin B secretion by lysosomes) as well. Dysregulation of the ability of lysosomes to fuse with autophagosomes,or dysregulation of their transport or maturation has huge implications in neuronal homeostasis including the ability to remodel the extracellular matrix to induce structural plasticity of axonal or synaptic structures.
Cathepsin B Secretion and Extracellular Matrix Remodeling to Promote Cell Migration
The role of cathepsins, especially cathepsin B, in terms of cell migration is best characterized in the tumor microenvironment as it relates to cancer cell metastasis (Olson and Joyce, 2015).For example, increased cathepsin expression is positively correlated with poor prognosis and increased metastasis in tumors such as breast, lung, and nasal pharyngeal cancers among others (Berdowska, 2004; Jedeszko and Sloane, 2004).Specifically, the role of cathepsin B in extracellular matrix degradation and the establishment of “invasion trails” upon which other invasive tumor cells can use as a guide to expand the tumor environment (Olson and Joyce, 2015) is well characterized. Use of the drug, CA074, to inhibit cathepsin B activity was reported to attenuate inflammatory breast cancer invasion through decreasing tumor cell migration across a more intact extracellular matrix (Victor et al., 2011).Additionally, cathepsin B plays a role in this process by cleaving cell-cell adhesion molecules (Sevenich and Joyce, 2014) and importantly, its secretion into the extracellular space degrades inhibitory extracellular matrix proteins to allow for disinhibited cell migration (Gocheva et al., 2010). Somewhat like cancer cells, adult axons attempting to sprout or regenerate following traumatic spinal cord injury apply these strategies(albeit inadequately) to navigate through an inhibitory post injury environment where, for example, CSPGs are highly upregulated along the glial scar and synapse-encapsulating structures called perineuronal nets (Tran et al., 2018b).
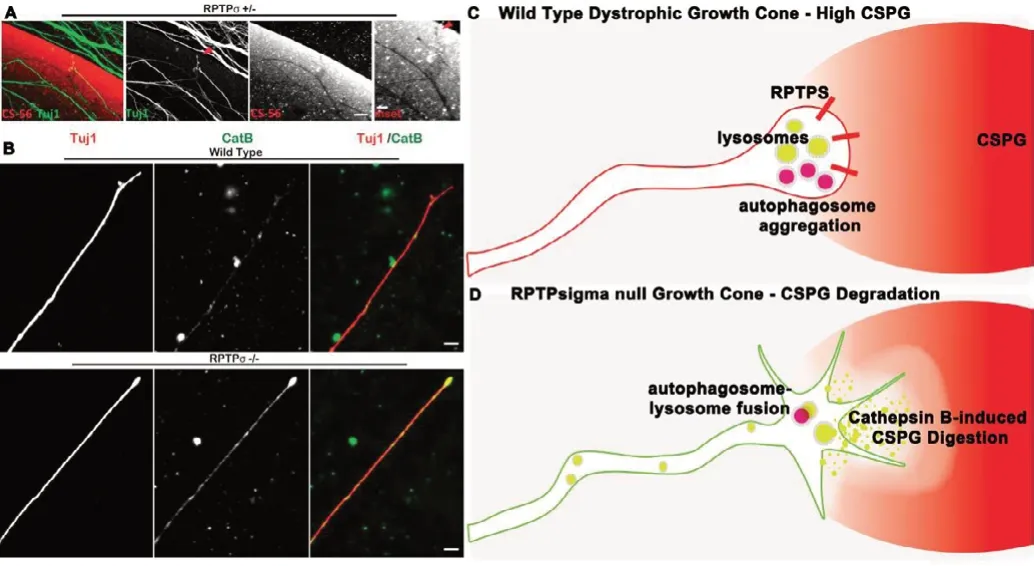
Figure 3 |Role of cathepsins in axon regeneration.Localized secretion of cathepsin B contributes to enhanced axon outgrowth.(A) Peripheral dorsal root ganglion neurons derived from RPTPsigma heterozygous mutants plated on axon-inhibitory chondroitin sulfate proteoglycans (CSPG) are able to cross this inhibitory gradient using localized secretion of Cathepsins B. Wild type peripheral neurons would otherwise become entrapped by the gradient of CSPGs. (B) RPTPsigma mutants show increased immunostaining of cathepsin B along the axon as well as the leading edge where it is well-positioned to digest inhibitory CSPGs. (C) The presence of transmembrane receptor PTPsigma in wild type axons binds to CSPGs causing aggregation of lysosomes and autophagosomes. This contributes to axon regeneration failure and results in a dystrophic morphology. (D) The lack of RPTPsigma relieves its usual inhibition of cathepsin B. Enhanced Cathepsin B secretion into the CSPG-rich extracellular space allows for immediate CSPG digestion. Through another pathway, the lack of RPTPsigma also results in enhanced autophagosome-lysosomal fusion resulting in enhanced autophagic flux. Together, these mechanisms encourage axon outgrowth despite the presence of CSPGs. Figure 3A and B was reprinted from Tran et al. (2018).
Neuronal growth-cone induced “trails” of digested extracellular matrix have been observedin vitrosince the 1980s by the Nicolas Seeds group (Krystosek and Seeds,1981; McGuire and Seeds, 1990). Recently, our group found that cathepsin B secretion by peripheral dorsal root ganglion neurons was necessary to produce CSPG-digestion trails needed for axons to navigate past an inhibitory CSPG gradientin vitro(Tran et al., 2018a). In Tran et al. (2018a)study, we found that modulation of PTPσ with a synthetic peptide previously found to induce serotonergic axon sprouting and functional locomotor and bladder recovery following contusive spinal cord injury was able to dramatically upregulate cathepsin B activity in peripheral neurons (Lang et al., 2015). Treatment of peripheral neuronsin vitrowith the synthetic peptide called ISP was able to up-regulate Exo1-dependent secretion of cathepsin B into the extracellular matrix to specifically degrade inhibitory CSPGs. ISP treatment also enhanced cathepsin B immunoreactivity in sprouting serotonergic neuronsin vivofollowing contusive spinal cord injury. Immunostaining for GAG-CSPGs and serotonin neurons following spinal cord injury showed a reciprocal patterning of decreased GAG-CSPG staining where serotonin sprouting was remarkably increased in ISP-treated spinal cords. In contrast,immunostaining in the vehicle control spinal cords showed a decrease in serotonergic sprouting and an increase in GAGCSPG expression within the perineuronal net. Considering recent findings by Sakamoto et al. (2019), it is possible that ISP-modulation of PTPσ may result in dis-inhibition of cortactin and subsequent lysosomal secretion of cathepsin B into the extracellular matrix. Thus, PTPσ may act as a sensor to detect a pro-regenerative environment (no CSPGs) or an inhibitory environment (abundant CSPGs) to regulate cathepsin B secretion downstream in an effort to facilitate axon outgrowth.
Interestingly, the intrinsic mechanism of cathepsin B-induced axon outgrowth mirrors a well-characterized therapeutic strategy used to experimentally induce axon regeneration/plasticity: use of chondroitinase ABC to degrade extracellular inhibitory CSPGs. Like cathepsin B, chondroitinase ABC is an enzyme capable of cleaving CPSGs. However, chondroitinase is bacterially-derived and only cleaves the inhibitory sugar moieties of CSPGs. Treatment with chondroitinase ABC was found repeatedly to enhance neuronal regeneration/plasticity in a wide variety of models (Bradbury et al., 2002; Alilain et al., 2011; Tran et al., 2018b; Warren et al., 2018). This strategy of protease-induced degradation of CSPGs in the extracellular matrix and subsequent disinhibition of neuronal plasticity is also seen in cathepsin induced synapse plasticity, discussed in the following section. Of course, cathepsins possess other strategies to promote axon outgrowth such as specifically cleaving secondary effectors or receptors such as SEZ6L2(Boonen et al., 2016), integrins (Jevnikar et al., 2011), and other proteins regulating cellular adhesion (Turk et al., 2012).These strategies, however, are best discussed elsewhere.
The Role of Cathepsins in Synaptic Plasticity
In this section, we will discuss the growing body of work linking cathepsins with synaptic plasticity. Broadly, synaptic plasticity is the cellular process by which patterns of synaptic activity may result in changes in synaptic strength that ultimately result in learning and memory (Langille and Brown,2018). Mechanisms that lead to variations in synaptic strength include alterations in specific gene expression patterns,transport of proteins into the axon or dendrite brought about by these gene changes, and remodeling of dendritic spines or axon terminal arbors (Bailey et al., 2015). Below, we will describe studies showing how cathepsin B contributes to long lasting pre- and post-synaptic structural changes, which ultimately serve to alter synaptic strength either positively or negatively.
Observations of mouse models genetically deficient in certain cathepsins have long been correlated with synaptic pathology. Constitutive knock-out of cathepsin D, for example,results in a mouse model of congenital neuronal ceroidlipofuscinosis, an aggressive neurodegenerative disease that leads to axonal degeneration, early death, and is notably marked by presynaptic abnormalities (Partanen et al., 2008;Koch et al., 2011). Indeed, knock-out of cathepsin D resulted in loss of synapses coupled with aggregation of pre-synaptic proteins including alpha-synuclein and SNARE proteins(Partanen et al., 2008; Cullen et al., 2009). Koch et al. (2011)also observed increases in docked synaptic vesicles and a decreased frequency of miniature excitatory postsynaptic currents in the CA1 region of the hippocampus of P16 and P24 cathepsin D knock-out mice. These researchers further suggested that these synaptic pathologies contributed to the initiation of synaptic disassembly and later, neuronal degeneration in this mouse model of congenital neuronal ceroid-lipofuscinosis. In mouse models of Alzheimer’s disease(and discussed above), compensatory drug-induced lysosomal upregulation of cathepsins B, D, and L in hippocampal slices was found to clear accumulated synaptic proteins such as APP fragments which, in turn, restored microtubule integrity and synaptic efficacy (Bendiske and Bahr, 2003). In N-methyl-D-aspartate-induced models of excitotoxicity in wild type rat hippocampal slices, inhibition of cathepsins and other proteases with the drug N-acetyl-leucyl-leucyl-norleucinal prevented activity dependent N-methyl-D-aspartate-induced post-synaptic density thickening (Fukunaga et al., 2015).Further work by Graber et al. (2004), suggests that cathepsins may be modulating spine morphology through proteolysis of F-actin and other substrates such as glutamate decarboxylase.Clearly, regulated cathepsin activity is important in synaptic homeostasis as dysregulated cathepsin expression is positively correlated with a variety of neurodegenerative diseases with synaptic pathologies.
In addition to maintaining homeostasis, cathepsins are becoming more recognized for the role they play in activitydependent synaptic plasticity (Figure 4). Dendritic spines are postsynaptic protrusions that receive inputs from other neurons (Nishiyama, 2019). Cathepsin B and L, for example,were observed to instigate spine collapse and eliminate synapses by degrading the actin crosslinking and plasma membrane protein MARCKS in N-methyl-D-aspartate-induced excitotoxity (Graber et al., 2004). Cathepsin S, additionally,when expressed by microglia was shown to mediate sleepdependent synapse refinement (Hayashi et al., 2013).Increasingly, the role of cathepsins and lysosomal exocytosis in pre- and post-synaptic growth is becoming more appreciated.In the post-synaptic dendritic spine, specifically, Padamsey et al. (2017) found that lysosomal exocytosis and subsequent release of cathepsin B maintained activity-dependent and long-lasting structural spine changes and growth. Structural changes in spines are an important hallmark of synaptic plasticity and ultimately a requirement for learning and memory as this mechanism allows for strengthening of specific neural circuits (Caroni et al., 2012; Nakahata and Yasuda,2018). In this way, architectural changes in spines allow for learning and memory to persist throughout development and adulthood. Padamsey et al. (2017) sought to examine the role of lysosomes in the dendritic spines of pyramidal hippocampal neurons in slices derived from rats around postnatal day 14. They found that back propagating action potentials and subsequent calcium signaling caused lysosomal fusion with the plasma membrane. A subsequent consequence of lysosomal fusion with the plasma membrane was the extracellular release and activation of cathepsin B, which then activated MMP-9 signaling (Figure 4). As mentioned in prior sections, cathepsin B is well known in the cancer literature to be a “hub-like” enzyme, which acts on MMP-9 among many proteases extracellularly (Olson and Joyce, 2015; Ruan et al., 2016). MMP-9, in particular, plays an important role in maintaining long-term potentiation and enhancing dendritic spine growth (Wang et al., 2008; Michaluk et al., 2011;Wlodarczyk et al., 2011). Such was the case in Padamsey et al. (2017) who found that cathepsin B release was necessary for MMP-9 mediated spine growth. Inhibition of cathepsin B with a relatively non-specific cathepsin B inhibitor, CA-074,ablated the ability of paired pulse stimulations to enhance spine growth. Simultaneous cathepsin B and MMP-9 release and activation in the extracellular environment may, in part,result in remodeling of the extracellular matrix (Padamsey et al., 2017). Degradation of inhibitory extracellular matrix proteins, such as CSPGs, for example, could further enhance and maintain long-lasting spine growth especially since CSPGs are a known target of cathepsin B (Fosang et al., 1992,1995; Tran et al., 2018a). Perisynaptic CSPGs, such as those accumulated in perineuronal nets surrounding the soma and proximal dendrites of select neurons, restricts structural plasticity (Frischknecht and Gundelfinger, 2012; Orlando et al.,2012) and enhances stability of neural circuits. CSPGs were shown to directly down-regulate spine dynamics in cortical neurons by specifically targeting tropomyosin-related kinase B protein (Kurihara and Yamashita, 2012). The adminstration of chondroitinase ABC enhanced spine motility and growth(Orlando et al., 2012).
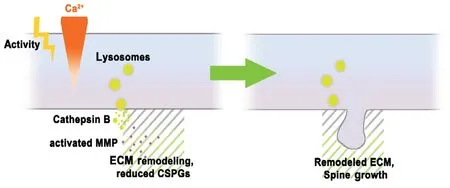
Figure 4 |The role of cathepsins in synaptic plasticity.Cathepsins contribute to structural synaptic plasticity by eliminating dendritic spines as well as inducing spine growth. In this model of activity-dependent synaptic plasticity, activity causes the intracellular increase of calcium, which induces lysosomal exocytosis and release of cathepsin B into the extracellular space. Cathepsin B activates matrix metalloproteinase (MMP-9) to induce local extracellular matrix (ECM) remodeling, including digestion of chondroitin sulfate proteoglycans (CSPGs). Remodeled ECM allows for enhanced dendritic spine growth and long-lasting structural changes. See Padamsey et al. (2017)and Ibata et al. (2019) for more details.
At the burgeoning pre-synapse formed by an axon approaching its post-synaptic target, cathepsin B exocytosis is also implicated in synapse modification. Ibata et al. (2019)found that a synaptic organizing protein, Cbln1, was cosecreted with cathepsin B from the lysosomes of pre-synaptic axonal boutons in cerebellar granule cells in an activity and calcium-dependent manner. The authors additionally found that Cbln1 diffused along the axon surface and bound to postsynaptic delta2 glutamate receptors in concentrated boutons to instigate synaptogenesis. Inhibition of axonal lysosomal exocytosis with Neu1, a sialidase, significantly reduced synaptogenesis (Ibata et al., 2019). In this case, it is also likely that simultaneous lysosomal exocytosis of cathepsin B with Cbln1 from axons serves to coordinate synapse formation with the postsynaptic dendrite by remodeling the extracellular matrix and inducing long-lasting structural changes (Figure 4). Together, these studies strongly suggest that cathepsin B plays an important role in the regulation and coordination of spine growth, motility and synaptogenesis by remodeling the extracellular matrix.
Conclusion
In order to stabilize lesions or protect synapses, inhibitory CSPGs are upregulated following spinal cord injury or traumatic brain injury, as well as a variety of neurodegenerative diseases such as Alzheimer’s disease (Morawski et al., 2014; Fawcett,2015) or multiple sclerosis (Keough et al., 2016; Luo et al.,2018) where chronic inflammatory processes continually provoke immune cells and reactive glia (Furman et al., 2019).Sometimes, drug-induced, controlled secretion of cathepsins enhanced plasticity and even restored function (Tran et al., 2018a). However, the uncontrolled upregulation and release of cathepsins induced tissue damage after traumatic injury to the CNS or by producing pathological forms of amyloid precursor protein in Alzheimer’s disease. How can cathepsins both enhance regeneration/plasticity and drive neurodegeneration? As we have discussed, cathepsins need to be heavily regulated at each level of their production and secretion. In the context of inflammation such as that which occurs immediately following CNS injury and in certain models of neurodegenerative disease, dysregulation of these regulatory processes may go awry. In response to intense inflammation and NF-kB or other cytokine signaling, for example, transcription and release of cathepsins may exceed that which can be endogenously controlled by cystatins leading eventually to compromised integrity of otherwise healthy cells. So, too, in spinal cord injury, the resulting protease storm induces secondary tissue damage and cell death at the lesion epicenter, further reducing function well beyond that which might have been produced by the initial mechanical trauma. Moving forward, it will be important to harness cathepsin activity in specific cell types as a promising therapeutic target to enhance neuronal plasticity after trauma or disease.
Author contributions:APT searched the literature, outlined and wrote the manuscript and created figures. JS edited and revised the manuscript.Both authors approved the final manuscript.
Conflicts of interest:A patent (9,937,242) has been granted for ISP, filed by Jerry Silver. The remaining author declares no competing interests.
Financial support:JS was funded by NINDS (NS25713), Brumagin-Nelson Fund, Kaneko Family Fund, and the Hong Kong Spinal Cord Injury Fund.
Copyright license agreement:The Copyright License Agreement has been signed by both authors before publication.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix,tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.

- 中國神經(jīng)再生研究(英文版)的其它文章
- Oxidative stress battles neuronal Bcl-xL in a fight to the death
- Development and postnatal neurogenesis in the retina:a comparison between altricial and precocial bird species
- The role of peptidase neurolysin in neuroprotection and neural repair after stroke
- Cognitive impairment in multiple sclerosis: lessons from cerebrospinal fluid biomarkers
- Progenies of NG2 glia: what do we learn from transgenic mouse models ?
- The NLRP3 inflammasome: a potential therapeutic target for traumatic brain injury