
The NLRP3 inflammasome: a potential therapeutic target for traumatic brain injury
2021-08-20 12:36SaifudeenIsmaelHebaAhmedTusitaAdrisKehkashanParveenParthThakorTauheedIshrat
中國(guó)神經(jīng)再生研究(英文版) 2021年1期
Saifudeen Ismael, Heba A. Ahmed, Tusita Adris, Kehkashan Parveen,Parth Thakor, Tauheed Ishrat,
Abstract Although the precise mechanisms contributing to secondary brain injury following traumatic brain injury are complex and obscure, a number of studies have demonstrated that inflammatory responses are an obvious and early feature in the pathogenesis of traumatic brain injury. Inflammasomes are multiprotein complexes that prompt the stimulation of caspase-1 and subsequently induce the maturation and secretion of proinflammatory cytokines, such as interleukin-1β and interleukin-18. These cytokines play a pivotal role in facilitating innate immune responses and inflammation. Among various inflammasome complexes, the NOD-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome is the best characterized, a crucial role for NLRP3 has been demonstrated in various brain diseases, including traumatic brain injury. Several recent studies have revealed the contribution of NLRP3 inflammasome in identifying cellular damage and stimulating inflammatory responses to aseptic tissue injury after traumatic brain injury. Even more important, blocking or inhibiting the activation of the NLRP3 inflammasome may have substantial potential to salvage tissue damage during traumatic brain injury. In this review, we summarize recently described mechanisms that are involved in the activation and regulation of the NLRP3 inflammasome. Moreover, we review the recent investigations on the contribution of the NLRP3 inflammasome in the pathophysiology of TBI, and current advances and challenges in potential NLRP3-targeted therapies. A significant contribution of NLRP3 inflammasome activation to traumatic brain injury implies that therapeutic approaches focused on targeting specific inflammasome components could significantly improve the traumatic brain injury outcomes.
Key Words: inflammation; interleukin-1β; NLRP3-inflammasome; caspase-1; MCC950;traumatic brain injury
Introduction
Traumatic brain injury (TBI) is one of the leading causes of mortality and disability across the globe, associated with life-threatening public health problems in people of all ages(Ghajar, 2000; Levin and Diaz-Arrastia, 2015). An estimated 2 million people suffer from TBI each year in the US, and, of those, 50,000 die and 80,000 sustain permanent disabilities(Lin et al., 2017). Additionally, there are over 60,000 children hospitalized because of TBI per year in the USA alone (Moreau et al., 2013). Many of those suffer from mild to moderate neurological deficits, which impair their lives irrespective of age or gender (Finnie, 2013). Based on the severity of injury, TBI can be mild, moderate or severe. The current management of TBI focuses on detection and treatment of associated secondary injury, since the damage due to the primary injury is considered to be irreversible (Seule et al.,2015). Primary injury results from the mechanical damage to neuronal tissue that occurs at the time of TBI, including contusion and hemorrhage as a result of shearing, tearing or stretching. Secondary injury then occurs over minutes to days and even months after the initial insult and is characterized by neurochemical, metabolic and behavioral alterations(Kumar and Loane, 2012). The consequences of brain injury can induce behavioral, emotional and cognitive deficiencies,and thus unfavorably affect the quality of life. Rapid and effective treatment is necessary to prevent undesirable complications and promote better recovery. Standard therapy is supportive in nature and focuses on the prevention of secondary complications. New research is ongoing in an effort to elucidate therapeutic strategies for attenuating the devastating consequences of TBI.
The pathomechanisms of TBI stem from mechanical damage that trigger multifactorial secondary cellular responses including disturbances in ionic homeostasis, glutamate excitotoxicity, mitochondrial dysfunction, oxidative stress, lipid degradation, diffused axonal injury, initiation of inflammatory and immune responses, and cellular apoptosis (Nizamutdinov and Shapiro, 2017). The initial insult triggers the invasion and migration of immune cells such as macrophages and neutrophils to the injured area, thereby inducing inflammation and edema. These neurochemical events induce the generation of pro-inflammatory mediators such as cytokines, chemokines and prostaglandins, ultimately resulting in blood-brain barrier(BBB) leakage and the development cerebral edema (Kumar and Loane, 2012). More than thirty prospective clinical trials for treating TBI have not shown significant promise, as these trials focused on single mechanisms of secondary injury while the mechanisms involved in TBI are clearly multifactorial in nature (Maas et al., 2010).
Although, the exact mechanisms triggering secondary brain injury remain ambiguous, it is suggested that inflammatory responses contribute to the pathophysiology. Post-traumatic neuroinflammation is a central response to the secondary injury that leads to continuous neurodegeneration and neurological impairment. It is characterized by microglial activation, leukocyte recruitment and secretion of inflammatory mediators (Morganti-Kossmann et al., 2007). Gene expression profile analyses in animal models of TBI demonstrated that genes related to inflammation and transcriptional regulators are strongly elevated in the acute phase of injury (Kobori et al.,2002; Natale et al., 2003). Rapid elevation of pro-inflammatory cytokines such as interleukin (IL)-1β, are observed within hours of TBI in both rodents and humans (Li et al., 2011; Lin et al.,2017). The secreted IL-1β can activate other pro-inflammatory cytokines, including tumor necrosis factor-α (TNF-α) which will further exacerbate the secondary tissue damage (Lu et al.,2005). IL-1β maturation (cleavage of proform) and its release to the systemic circulation is predominantly regulated by the NLRP3 inflammasome, via a newly described mechanism that governs inflammatory responses during secondary tissue injury (Henao-Mejia et al., 2012; Karmakar et al., 2015). Mainly localized in immune cells, the nucleotide oligomerization-like receptor protein 3 (NLRP3) inflammasome is composed of NLRP3, oligomers apoptosis-associated speck-like (ASC) adapter protein and pro-caspase-1, serving as a platform for cleavage of caspase-1 and subsequent IL-1β maturation. According to recent findings, TBI appears to coincide with NLRP3 and ASC oligomerization, inducing the activation of caspase-1 and subsequent IL-1β and IL-18 maturation (Figure 1) (Liu et al.,2013; Fan et al., 2017). This finding may be highly significant since NLRP3 has been suggested as a cerebrospinal fluid (CSF)biomarker in TBI patients with poor prognosis (Wallisch et al., 2017). Despite all this evolving research, little has been evaluated with regards to the potential benefit of NLRP3 inhibitors in TBI. Notably, recent studies from our groups and others indicated that pharmacological inhibition of NLRP3 improves neurological outcomes in animal models of TBI. The present review focuses on the mechanism of the inflammatory responses, mediated by the NLRP3 inflammasome and its possible therapeutic potential in the management of TBI.
Search Strategy and Selection Criteria
The most relevant publications included in this article were identified in the PubMed database: https://www.ncbi.nlm.nih.gov/pubmed/. 1) Keywords used for searching (selection criteria): traumatic brain injury, NLRP3 inflammasome, brain,inhibitors, inflammation; neurodegeneration. 2) Dates of searching: 2000-2019.
The Nucleotide Oligomerization-Like Receptor Protein 3 Inflammasome
The innate immune response to cellular stress, infection andtissue injury occurs via pattern- recognition receptors (PRRs)expressed mainly by inflammatory cells such as monocytes,macrophages, neutrophils, and “inflammatory” dendritic cells. PRRs are components of innate immune system, which recognize pathogen-associated molecular patterns (PAMPs),of pathogens, and damage-associated molecular patterns(DAMPs), components of host cells that are released during cellular damage (Schroder and Tschopp, 2010). Toll-like receptors prime the signaling cascade that leads to cellular activation and production of inflammatory mediators, such as TNF, IL-6, IL-8 and type I interferons (IFNs), in response to extracellular signals. Nod- like receptors (NLRs), a group of PPRs, function in the response to danger signals introduced into the host cell cytoplasm. There are more than 20 NLR genes in humans and more than 30 in mice (Ting et al., 2008).These NLRs include NLRP1, NLRP3, NLRC4, NLRC5, NLRP6,NLRP7, NLRP12, as well as the non-NLR inflammasome receptor known as AIM2. Among these, NLRP3 is the most studied inflammasome.
The NLRP3 inflammasome was first described in the Muckle-Wells autoinflammatory disorder, which is associated with spontaneous release of IL-1β (Agostini et al., 2004). The NLRP3 inflammasome is activated by various stimuli and stimulates the activation of caspase-1 (the effector molecule), which sequentially leads to the maturation and release of IL-1β and IL-18, and ultimately potentiates inflammatory responses(Agostini et al., 2004; Sutterwala et al., 2006). NLRP3 contains an amino-terminal-terminal pyrin-only domain (PYD), a conserved central nucleotide binding and oligomerization domain (nucleotide oligomerization or NACHT) and a carboxyterminal leucine-rich repeat (LRR) domain (Jo et al., 2016).The LRR domain senses PAMPs and other ligands, maintains the NLRP3 in active state, and facilitates protein-protein interactions. The NACHT domain binds with nucleotides, and is vital for protein oligomerization of NLRP3 inflammasome assembly process (Duncan et al., 2007). The PYD domain supports the binding between NLRP and the bipartite adapter ASC via homophilic interactions resulting in the large speck-like structure (Liepinsh et al., 2003). ASC consists of an N-terminal PYD and a C-terminal caspase activation and recruitment domain (CARD). ASC communicate with procasapase-1 through the CARD domain (Srinivasula et al.,2002). Under resting state caspase-1 exists as a catalytically dormant precursor, pro-caspase-1. Induction of pro-caspase-1,contributes to the proteolytic maturation of the proenzyme into its active form (Agostini et al., 2004). The activated caspase-1 further induces the maturation and release of proinflammatory cytokines IL-1β and IL-18, which belong to the IL-1 family and mediate succeeding immune responses (Keller et al., 2008).
The role of mature caspase-1 is not restricted to secretion of proinflammatory mediators, in fact activated caspase-1 induces inherent inflammatory cell death, known as pyroptosis, which is characterized by cellular swelling, rapid release of intra cellular contents and DNA fragmentation(Bergsbaken et al., 2009; Liu et al., 2018a). Additionally,rupture of the plasma membrane is followed by excessive secretion of pro-inflammatory mediators such as TNF-α, IL-1β, IL-6 and CX3C-chemokine ligand 3, which may exacerbate pyroptosis as is observed during the acute phase of TBI (Fink et al., 2008; Liu et al., 2018a). Pyroptosis is also known as caspase-1 dependent cell death, and is morphologically and mechanistically distinct from apoptosis and other forms of cell death (Bergsbaken et al., 2009).
Regulation of Nucleotide Oligomerization-Like Receptor Protein 3 Inflammasome Activation
The NLRP3 inflammasome plays a crucial role in priming inflammatory responses. NLRP3 is induced by a variety of stimuli including pore-forming toxins, extracellular ATP in the presence of various pathogen-associated molecules,uric acid crystals, virus-associated DNA, RNA, asbestos and ultraviolet-B irradiation (Gurcel et al., 2006; Kanneganti et al.,2006; Mariathasan et al., 2006; Martinon et al., 2006; Koo et al., 2008; Muruve et al., 2008). However, the mechanism by which NLRP3 responds to multiple stimuli is elusive. ATP acts as an extra cellular NLRP3 agonist, inducing activation and oligomerization of the inflammasome by inducing K+efflux through a P2X purinergic receptor 7 (P2X7)-dependent pore consisting of a pannexin-1 hemichannel. Additionally, NLRP3 can be induced by changes in cellular ionic homeostasis such as low K+concentration, Ca2+flux, redox imbalance or mitochondrial dysfunction (Pétrilli et al., 2007; Zhou et al.,2010, 2011; Lee et al., 2012). Several PAMPs and DAMPs induce the activation of the NLRP3 inflammasome, mediated via the thioredoxin-interacting protein (TXNIP) (van Bruggen et al., 2010). Mitochondrial reactive oxygen radicals are the primary stimuli for activation of TXNIP (Kim et al., 2012), and interactions between TXNIP and NLRP3 stimulate the secretion of inflammatory mediators (Zhang et al., 2015). Cytosolic NADPH oxidase 2 (NOX2) also contributes to the activation of the NLRP3 inflammasome through production of reactive oxygen species (Abais et al., 2015). NLRP3 is negatively regulated by nuclear factor erythroid-2 related factor 2(Nrf2), a redox sensitive transcription factor that regulates expression of cellular antioxidant system. Activators of Nrf2 stimulates the transcription of cellular redox responses that scavenge oxidative damage and results in reduced priming of NF-κB (Jhang and Yen, 2017; Hou et al., 2018). Reduced Nrf2 activity is associated elevated intracellular reactive oxygen species (ROS) level eventually leads to dissociation of TXNIP from thioredoxin and increases the binding of latter to NLRP3(Jhang and Yen, 2017). Additionally, lysosome destabilization stimulates the activation of NLRP3 by the release of cathepsin B from its contents (Bruchard et al., 2013). Conversely,autophagy acts as negative controller of NLRP3 inflammasome activation (Figure 1) (Nakahira et al., 2011).
The intrinsic activity of NLRP3 is modulated by distinct mechanisms. BRCA1-BRCA2-containing complex-3(deubiquitinating enzyme), double-stranded RNA-dependent protein kinase, death-associated protein kinase 1 and Bruton’s tyrosine kinase function as positive regulators of NLRP3 inflammasome activity (Song et al., 2017). A member of the NIMA-related kinase (NEK) family, NEK7 has been proven to directly interact to the LRR domain of NLRP3 and act downstream of K+efflux and ROS generation to stimulate the oligomerization of NLRP3 inflammasome (He et al., 2016). K+efflux triggers NEK7-NLRP3 assembly which leads to activation of NLRP3 inflammasome, cleavage of pro-caspase-1, and subsequent neuronal injury. NEK7 knockdown inin vivoexperiments reduces NEK7-NLRP3 binding and attenuates NLRP3 inflammasome activity, pro-caspase recruitment, and pyroptosis in nerve injuries status- post TBI. Additionally,autophagy, microRNAs, CARD-only proteins, pyrin-only proteins and nitric oxide (NO) act as endogenous negative regulators of NLRP3 (Saitoh et al., 2008; Hernandez-Cuellar et al., 2012; Yang et al., 2015; de Almeida et al., 2015).
Role of the Nucleotide Oligomerization-Like Receptor Protein 3 Inflammasome in Traumatic Brain Injury
The main mechanism of secondary brain injury following TBI is known as perilesional edema and is characterized by increased secretion of pro-inflammatory markers, recruitment of innate immune cells into the brain, and activation of local brain astrocytes and microglia (Yi et al., 2019). Accumulating evidence indicates that activation of the NLRP3 inflammasome occurs during the secondary injury of TBI, this has shed a new light on understanding the pathophysiology and development of new strategies for the management of TBI. A temporal relationship between the expression of the NLRP3 inflammasome components and TBI has been reported(Liu et al., 2013). Injury initiated elevated expression of components of the NLRP3-inflammasome, up-regulation of the ASC and caspase-1, and led to maturation and cleavage of IL-1β and IL-18. At the cellular level, NLRP3-inflammasome was identified in neurons, astrocytes, and microglia in the pericontusional cortex. There was a drastic increase in the IL-1β secretion in the pericontusional area at 6 hours post-injury,with a rapid decrease beginning at 24 hours. Conversely, IL-18 protein expression was elevated gradually from 6 hours to 7 days suggesting a role in delayed injury. Supporting data reported a delayed secretion of IL-18 following TBI occurred,which gradually increased over a period of 7 days (Qian et al., 2017). Moreover, protein expression of NLRP3, ASC and caspase-1 continued to increase from 6 hours to 7 days.Acute stimulation of NLRP3 inflammasome components were also observed in a rodent model of penetrating ballisticlike brain injury, which simulates cranial gunshot injury and is associated with progressive tissue loss (Lee et al., 2018).NLRP3 inflammasome activation reached peak at 48 hours after injury followed by the induction of pyroptosis. Initially the inflammasome activation was localized predominantly in neurons and later switched to microglia between 24 and 48 hours after injury. This could be explained by the pyroptotic response of neurons, later stimulate inflammasome activation in neighboring microglia, whereas astrocytes showed delayed inflammasome activation (Lee et al., 2018). At 48 hours after penetrating ballistic-like brain injury, there was a significant rise in the population of primed and ameboid-type microglia expressing inflammasome proteins. These microglia persisted in the injured brain for up to 12 weeks post-injury with continuous neurodegeneration. The possible reason for the discrepancy in the temporal profile of inflammasome expression may be due to variability in extent of brain jury and difference in animal model. The temporal expression pattern of NLRP3 inflammasome components implies that, initial cell death induced by mechanical injury stimulate innate immune response by rapid elevation of NLRP3 and later it is down regulated by activation of protective mechanisms of the body.
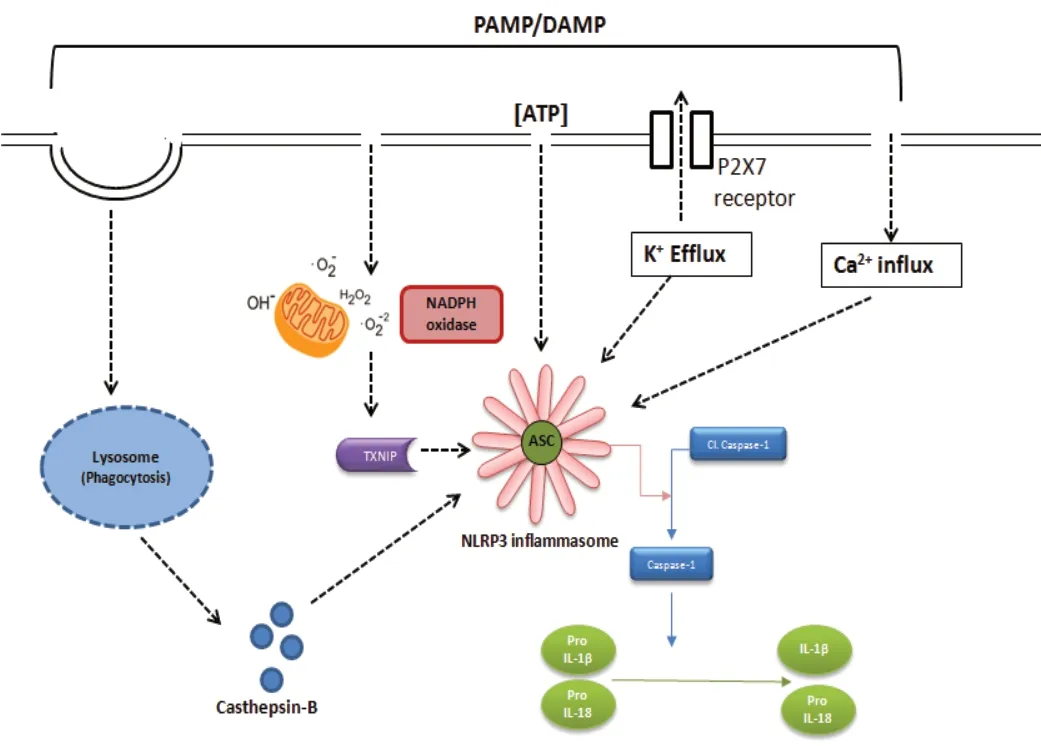
Figure 1 |Factors influencing the assembly and activation of the NLRP3 inflammasome.NLRP3 is activated by a number of different regulators including various PAMPs and DAMPs. NLRP3 is induced by disturbances in cellular ionic homeostasis such as K+ efflux and Ca2+ influx. Assembly of the NLRP3 inflammasome is mediated by the generation of reactive oxygen free radicals derived from mitochondrial and cytosolic NADPH oxidase. Activated ROS can induce TXNIP, and the interaction between TXNIP and NLRP3 contribute to the activation of NLRP3 inflammasome and the resulting release of inflammatory mediators. Destabilization of phagolysosomes releases lysosomal contents such as cathepsin B, which stimulates the activation of NLRP3. ASC: Apoptosisassociated speck-like protein; DAMP: damage-associated molecular patterns;IL-1β: interleukin-1 beta; IL-18: interleukin-18; NADPH: nicotinamide adenine dinucleotide phosphate; NLRP3: nucleotide oligomerization-like receptor protein 3; PAMP: pathogen-associated molecular patterns; P2RX7: P2X purinoceptor 7; TXNIP: thioredoxin interacting protein.
One study demonstrated the activation of NLRP3 inflammasome in mediating inflammatory responses in rodent model of blast induced TBI (Ma et al., 2016). They demonstrated that there was increased expression of thioredoxin-interacting protein(TXNIP), a redox sensitive critical regulator of NLRP3 activity,with elevated expression of NLRP3 and activation of caspase-1.
Additionally, there was a higher expression of proinflammatory cytokines including IL-1β and TNF-α in response to blast injury. Administration of propofol (2,6-diisopropyl phenol),a well-known lipid-soluble intravenous anesthetic, was accompanied by attenuation of NLRP3 expression, IL-1β maturation and oxidative stress. Similarly, hyperbaric oxygen treatment attenuated neuroinflammation and brain edema,with accompanied improvements in motor function (Qian et al., 2017). However, the beneficial effect of hyperbaric oxygen treatment on NLRP3 is elusive as it can induce oxidative stress which is known to induce NLRP3 inflammasome (Tschopp and Schroder, 2010). Additionally, the role of NLRP3 inflammasomeassociated IL-1β secretion in traumatic cerebral edema has been demonstrated (Erdi et al., 2016). Moreover, treatment with telmisartan, an angiotensin receptor blocker, ameliorated cerebral edema, and subsequently improved neurological outcome which was associated with decreased NLRP3 inflammasome assembly after TBI. The beneficial effect of telmisartan is mediated by inhibition of NLRP3 dependent IL-18 and IL-1β release (Wei et al., 2016). Elevated NLRP3 concentration was found in CSF of children with severe TBI.Elevated expression of NLRP1 was detected in only 11% of children with severe TBI in compared with uninjured controls(Wallisch et al., 2017).
Blast induced TBI is a form of brain trauma due to the use of explosives, for example those associated with war zones and terrorist activities. The resulting secondary damage induces free radical induced TXNIP expression and its downstream NLRP3 inflammasome activation (Fan et al., 2017). Treatment with mangiferin (1,3,6,7-tetrahydroxyxanthone-C2-β-Dglucoside), a component of traditional Chinese medicine,alleviated brain damage by inhibition of ROS-TXNIP-NLRP3 inflammasome pathway. Supportingly, the contribution of oxidative stress in mediating NLRP3 inflammasome stimulation was recently confirmed. The deletion of NOX-2, a component of NADPH oxidase, a major ROS inducer,attenuated NLRP3 inflammasome activation following TBI and was correlated with reduced interaction between TXNIP and NLRP3. NOX is a family of enzymes particularly devoted to the production ROS. Clinical (autopsy) and experimental studies have confirmed the elevated expression of NOX isoforms following TBI (Zhang et al., 2012; Li et al., 2015).This was localized cortex and hippocampus in animal models (Zhang et al., 2012). Additionally, pharmacological inhibition of NOX is associated with attenuation of apoptosis,inflammation and reduced TXNIP expression (Lucke-Wold et al., 2015). Enhanced oxidative stress, brain water contents(edema) and inflammatory markers that are associated with TBI were attenuated by treatment with resveratrol(Zou et al., 2018). TBI induces down regulation of Nrf2 and subsequent elevation of oxidative and nitrosative stress,which activates neuroinflammation and cellular apoptosis.Deletion Nrf2 function in mice resulted in exacerbated brain damage and inflammatory response in TBI model of fluid percussion injury (Bhowmick et al., 2019). These observations collectively demonstrated that modulation of oxidative stress may attenuate the TXNIP-NLRP3 inflammasome mediated secondary injury after TBI.
Later, we all got up and sat around the tree and opened the few wrapped presents. Afterward9 the children were given their three envelopes. We read the words with teary eyes and red noses. Then we got to “the baby of the family’s” notes. Erik, at 8, wasn’t expecting to hear anything nice. His brother had written: “What I love about my brother Erik is that he’s not afraid of anything,” Mia had written, “What I love about my brother Erik is he can talk to anybody!” Lisa had written, “What I love about my brother Erik is he can climb trees higher than anyone!”
Recently, activation of NLRP3 inflammasome was also reported in a rodent model of controlled cortical impact injury(CCI). In this study, treatment with ω?3 fatty acids, alleviated inflammation and behavioral deficits. This response was mediated by the elevated expression of anti-inflammatory β-Arrestin-2 scaffold protein, a downstream molecule of GPR40, which negatively modulates NLRP3 inflammasome activation. The role of NLRP3 has been further confirmed by both genetic and pharmacological approaches. NLRP3 knock out mice demonstrated a preserved cognitive function, less severe damage and less inflammatory response compared to wild-type mice in response to TBI (Irrera et al., 2017).
Additionally, pharmacological inhibition of NLRP3 with BAY 11-7082 revealed more preserved brain structure and reduced edema following TBI. Recently, we have reported that the inhibition of NLRP3 inflammasome components protected mice from TBI induced brain damage in a murine model of CCI(Ismael et al., 2018a). Pharmacological inhibition with highly specific small molecular inhibitor MCC950 protected mice from edema, and resulted in improved neurological function.The study validated both the acute (24 hours) and subacute(72 hours) phases after TBI. Treatment with MCC950 reduced the level of apoptotic markers such as cleaved caspase-3 and cleaved poly (ADP-ribose) polymerase (PARP), while causing an upregulation of cell survival molecule protein kinase B.MCC950 also attenuated NLRP3 inflammasome priming by modulation of NFκB p65 expression. However, the study was unable to validate the long term beneficial effect on neurological outcome and structural integrity of the damaged brain. Similar studies were later conducted by another group over an extended period and found elevated NLRP3 inflammasome expression in the peri-contusional cortex and microglia to be the primary source of NLRP3 expression (Xu et al., 2018). Inhibition of NLRP3 inflammasome activation is associated with an improved long term neurological outcome,preserved blood brain barrier integrity, reduced microglial activation, and leukocyte infiltration following TBI. However,MCC950 administration started 12 hours post-TBI did not result in any significant progress. The beneficial effects were evident up to 6 hous post TBI, suggesting that the therapeutic window for MCC950 may be 6 hours. Additionally, it was reported that α2-adrenergic receptor agonist that has been used as sedative hypnotic, analgesic (Fang et al., 2015) has beneficial effects in TBI via NFκB-mediated inhibition of NLRP3 inflammasome, and can attenuate neurological dysfunction(Wang et al., 2018). Dexmedetomidine, a novel α2-adrenergic receptor agonist, has been shown to alleviate post traumatic inflammation through inhibition of the NLRP3 inflammasome.Dexmedetomidine reduced BBB disruption and cellular apoptosis, and improved neurobehavioral outcomes in the acute phase of TBI (Wang et al., 2018). A recent study evaluated the potential role of receptor-interacting protein 3 (RIP3) in the pathogenesis of TBI (Liu et al., 2018b). RIP3 was found to be upregulated following TBI in parallel with neuroinflammation, impaired neurological and behavioral function. RIP3 ablation improved neurological function and alleviated NLRP3 inflammasome activation, suggesting it may have a regulatory role on the NLRP3 inflammasome.More recently it was demonstrated that NIMA-related kinase 7 (NEK7) mediated NLRP3 inflammasome activation and silencing of NEK7 attenuates NLRP3 inflammasome activation,neurological deficit and pyroptosis following TBI in mouse model of CCI. NEK7 shows a low level of activity under normal circumstances and increased in response to TBI, suggesting its role as the upstream instigator (He et al., 2016). The mechanism of activation of NLRP3 inflammasome in TBI is depicted inFigure 2.
TBI is a heterogeneous condition and may result from many different types of insults leading to variable inflammatory responses. There were differences in the temporal profiles of inflammasome activation between the various TBI models,these are likely corresponding to model-specific responses and tissue vulnerability patterns produced by the different models. This makes the comparison more difficult to interpret and limits its translation into clinical trials. Most of the preclinical studies for TBI are well monitored rapid studies,and none of the models simulate the exact clinical condition.Although several pharmacological agents have been validated in experimental studies, none of them have Food and Drug Administration approval for the treatment of TBI. There is an urgent need of specific therapeutic approach targeting multiple mechanism of disease progression in TBI.
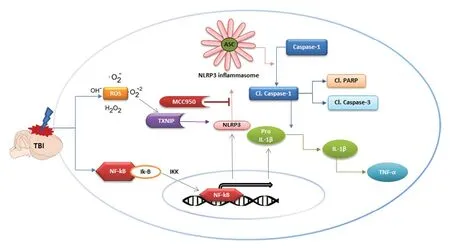
Figure 2 |Mechanism of activation of the NLRP3 inflammasome in TBI, and proposed mode of action of small molecule inhibitor MCC950.TBI activates the NLRP3 inflammasome by two different mechanisms, first the initial priming signal at the transcriptional level which induces the NF-κB pathway. Secondly, the induction mechanism involving the assembly of the NLRP3 inflammasome complex by direct interaction with TXNIP, a Redoxsensitive regulator. Activated NLRP3 induces the oligomerization of ASC which eventually leads to the assembly of the NLRP3 inflammasome complex.The assembled complex then catalyses the cleavage of Caspase-1 and the subsequent release of IL-1β and inflammatory mediators. Prolonged NLRP3 activation induces proapoptotic molecules such as cleaved PARP and cleaved caspase-3. The specific inhibitor MCC950 blocks NLRP3-induced ASC oligomerization, eventually alleviating the NLRP3-induced neuroinflammation and proapoptotic signaling caused by TBI. ASC: Apoptosis-associated speck-like protein; ATP: adenosine triphosphate; DAMP: damage-associated molecular patterns; IL-1β: interleukin-1 beta; IL-18: interleukin-18; NADPH: nicotinamide adenine dinucleotide phosphate; NLRP3: nucleotide oligomerization-like receptor protein 3; PAMP: pathogen-associated molecular patterns; P2RX7:P2X purinoceptor 7; TXNIP: thioredoxin interacting protein.
Role of Nucleotide Oligomerization-Like Receptor Protein 3 Inflammasome in Other Neurological Diseases and Conditions
NLRP3 mediated inflammasome activation is a pivotal signaling pathway in the innate immune system of the central nervous system (CNS). However aberrant activation of NLRP3 inflammasome signaling has been reported in the pathology of a wide range of neurological diseases. Clinical andin vivostudies reported the role of NLRP3 inflammasome in a broad spectrum of microbial infections of the CNS including pneumococcal meningitis,Staphylococcus aureusinfection (Hanamsagar et al., 2011; Hoegen et al., 2011).Moreover, aberrant expression of NLRP3 inflammosome has been described in viral infections with Japanese Encephalitis virus, influenza virus and human immunodeficiency virus(Hanamsagar et al., 2011; Hoegen et al., 2011; Kaushik et al.,2012; Walsh et al., 2014).
Several studies have validated the pathogenic role of the inflammasome in neurovascular diseases such as Alzheimer’s disease (AD), amylotropic lateral sclerosis, multiple sclerosis,ischemic stroke, traumatic injury and microbial infections(Kaushik et al., 2012; Inoue et al., 2012; Geldhoff et al.,2013; Liu et al., 2013; Zhao et al., 2015; Lammerding et al.,2016; Saresella et al., 2016). Inflammatory responses are essential for eliminating invading agents, clearing injured cells and promoting tissue repair. However, prolonged neuroinflammation may lead to further tissue damage and neurovascular dysfunction. The aberrant NLRP3 inflammasome activation is involved in many chronic inflammatory diseases including Alzheimer’s disease and aging, which is different from acute neuro inflammation. Chronic inflammation is a low-grade, controlled, chronic systemic inflammatory state and usually asymptomatic, where NLRP3 inflammasome is triggered by metabolic end by products such as urate and cholesterol crystals (Ozaki et al., 2015). In Alzheimer’s disease,the increase in Aβ plaques leads to release of DAMPS from neurons. These DAMPs are sensed by NLRP3 inflammasome complex leading to a cascade of events ending in prolonged neuroinflammation (Katsumoto et al., 2018). However, in the case acute brain injury such as TBI and stroke, DAMPs released from injured and necrotic cells and trigger the NF-κB and MAPK signaling pathway through PRRs. These activated pathway contribute to the expression and activation of NLRP3 inflammasome (Alishahi et al., 2019).
Recently we have demonstrated TXNIP mediated enhanced NLRP3 inflammasome activation in cerebral cortex of human AD brain (Li et al., 2019). Increased activation of NLRP3 was reported in intracerebral hemorrhage (ICH) and subarachnoid hemorrhage (SAH), characterized by enhanced production of inflammatory cytokines mediated by reactive oxygen free radicles and the P2X7R pathway (Wang and Doré, 2007; Keep et al., 2012; Feng et al., 2015). Ourin vivostudies showed that NLRP3 protein expression was found to be increased after experimental ischemic stroke, which was concomitant with an elevated expression of inflammatory mediators IL-1β and IL-18. Pharmacological inhibition of NLRP3 inflammasome improved cerebral ischemia outcomes, as demonstrated by reduced infarction volumes, alleviated neurovascular damage,and the attenuation of inflammatory markers (Ismael et al.,2018b). The significant contribution of NLRP3 in ischemic stroke and spinacord injury were extensively reviewed(Mortezaee et al., 2018; Alishahi et al., 2019).
Nucleotide Oligomerization-Like Receptor Protein 3 Inhibitors in Traumatic Brain Injury
A large number of investigations are warranted for developing a specific NLRP3 inhibitor for the treatment of TBI. Propofol and mangiferin were shown to provide significant inhibition of NLRP3 which is claimed to be responsible for their therapeutic activities in TBI (Fan et al., 2017). Telmisartan, an angiotensin receptor blocker, has also been reported to have NLPR3 antagonistic activity in the state of TBI (Erdi et al., 2016).One recentin vivostudy demonstrated that BAY11-7082 can attenuate NLRP3 action in the treatment of TBI (Irrera et al., 2017). Treatment with BAY 11-7082 led to a decrease in edema and preserved brain structure in a mouse model of TBI, even though BAY 11-7082 is not a direct inhibitor of NLRP3.
Hyperbaric oxygen treatment inhibited the production of inflammatory markers via inhibition of the NLRP3 inflammasome, but the exact mechanism still needs to be elucidated (Qian et al., 2017). Resveratrol, a nutraceutical drug, also alleviated brain damage following TBI by modifying the expression of NLRP3 inflammasome components,ROS and edema (Zou et al., 2018). The beneficial effect of resveratrol is mediated by NLRP3 attenuation via sirtuin 1 and ROS dependent mechanism. Previously, we have reported that resveratrol inhibits NLRP3 expression through a TXNIP mediated mechanism (Ishrat et al., 2015).
Artesunate, a derivative of the traditional Chinese phytochemical artemisinin that has been shown to have multiple pharmacological actions including antineuroinflammatory activity, administration of artesunate decreased the degree of brain injury and lesion volume by inhibiting NLRP3 inflammsome activation and NFκB nuclear translocation in mouse model of CCI (Gugliandolo et al.,2018). Additionally, artesunate protected agaist apoptosis,microglial activation, neuroinflammation and modulated the release of neurotrophic factors. Dexmedetomidine is an alpha 2 adrenergic receptor agonist whose administration in rats subjected to CCI inhibited NLRP3 inflammasome/capasase-1 axis activation, increasing neuronal viability, preventing microglial activation and improving overall cognitive function(Zheng et al., 2018). These responses were comparable to those seen with specific NLRP3 inhibitor BAY-11-7082 in TBI and showed synergetic effect on inhibiting NLRP3 inflammasome activation.
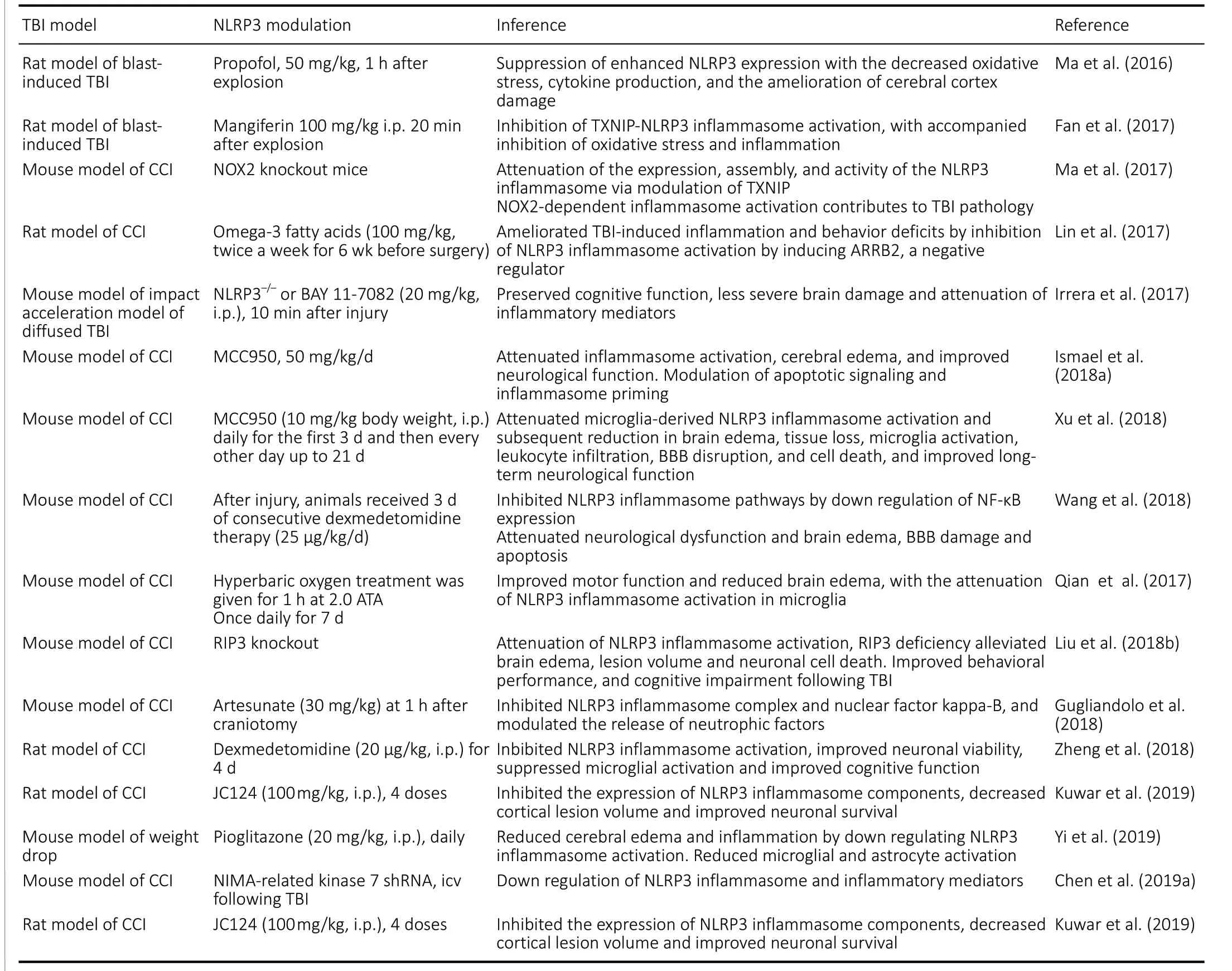
Table 1 |Summary of studies on the modulation of the NLRP3 inflammasome in TBI
Pioglitazone is a peroxisome proliferator-activated receptor agonist, belongs to the class of thiazolidinediones, a class of drugs used in the management of type 2 diabetes mellitus as it plays a role in increasing insulin sensitivity and promoting adipogenesis and fatty acid uptake. Pioglitazone administration following TBI significantly reduced perilesional cerebral edema through attenuation of NLRP-3 activity and subsequent downstream cytokines and in reducing activation of astrocytes and microglia (Yi et al., 2019).Table 1summarizes the studies on modulation of NLRP3 inflammasome in animal models of TBI.
MCC950, a Novel Selective Nucleotide Oligomerization-Like Receptor Protein 3 Inhibitor
MCC950 is a selective small molecule inhibitor of the NLRP3 inflammasome, and a promising candidate for the treatment of inflammatory diseases (Salla et al., 2016). It is a diarylsulphonylurea-containing compound, specific for NLRP3,but not for AIM2, NLRC4 or NLRP1 inflammasomes, and it blocks NLRP3-induced ASC oligomerization (Coll et al., 2015).
Because of its specificity, MCC950 is less immunosuppressive than other inflammasome inhibitors (Ren et al., 2018) and is orally bioavailable. The beneficial effects of MCC950 have been shown in experimental autoimmune encephalomyelitis and AD, where its ability to cross BBB was verified (Coll et al., 2015; Dempsey et al., 2017). Recently we demonstrated that inhibition of NLRP3 with MCC950 protect mice against TBI. This is the first report validating efficacy of MCC950 in a mouse model of TBI induced by controlled cortical impact (Ismael et al., 2018a). TBI induced activation of inflammasome, resulted in cerebral edema and impaired neurological function. MCC950 significantly alleviated the expression of inflammasome components such as NLRP3,ASC, cleaved caspase-1 and cleaved IL-1β. It was accompanied with improved neurological function and edema. Inhibition of NLRP3 attenuated the expression of other inflammatory mediator such as NFκB and TNFα. Additionally, MCC950 also provided protection against pyroptotic activation of PARP and caspase-3. Treatment with MCC950 modulated upstream NLRP3 regulator TXNIP. This was the first report addressing the beneficial effects of the selective NLRP3 inhibitor MCC950 on TBI, it was later validated that MCC950 has long term benefit in the mouse model of CCI (Xu et al.,2018). MCC950 attenuated NLRP3 inflammasome activation in the pericontusional area. It reduced brain edema and lesion volume in addition to improved long-term motor and cognitive functions with an activity window of 6 hours. A recent study reported that MCC950 reduces intracerebral hemorrhage via inhibition of the NLRP3 inflammasome (Ren et al., 2018).MCC950 reduced inflammation and attenuated neurological deficits and brain edema in mice after intracerebral hemorrhage was induced by injecting autologous blood or collagenase. Interestingly, treatment with MCC950 also enhanced BBB integrity and attenuated cell death. Recently,Chen et al. (2019b) demonstrated that NLRP3 inhibition along with enhancement of mitophagy with activation of mTOR(mammalian target of rapamycin) with rapamycin improves the neuroprotective effect of NLRP3 inflammasone inhibition following TBI. The proposed mode of action of MCC950 in TBI is depicted inFigure 2.
Recently, we have validated the potential benefit of MCC950 in the treatment of secondary brain damage following ischemic/reperfusion injury (Ismael et al., 2018b). Treatment with MCC950 decreased infarct size, edema, hemorrhage, the level of NLRP3 inflammasome components, and alleviated behavioral deficits in a mouse model of middle cerebral artery occlusion. Further long-term studies are required to validate these effects on long term behavioral outcomes. Although pre-clinical studies have validated the therapeutic potential of MCC950 in TBI, there is currently no clinical evidence in this regard. Hence, MCC950 may be a promising pharmaceutical approach for the treatment of TBI in the future.
Glyburide, a sulfonylurea derivative used in management of diabetes mellitus has shown NLRP3 inhibitory action in animal models, however its use is associated with development of hypoglycemia. Randomized clinical trial in patients with moderate and severe TBI shown that oral administration of glyburide is associated with decreased contusion expansion rate and improved neurological outcome (Zafardoost et al., 2016; Khalili et al., 2017). Additionally, intravenous administration of glyburide at low dose in marginally ameliorated lesion volume moderate to severe TBI patients(Eisenberg et al., 2019). Kuwar et al. (2019) has developed a novel small molecule inhibitor JC124 with high specificity for NLRP3 inflammasone. JC124 is structural optimization of glyburide to eliminate hypoglycemic effect. JC124 acts as an NLRP3 inhibitor by blocking ASC aggregation, activation of caspase-1 and release of IL-1β in macrophages and has been shown in rat brain injury models to be neuroprotective in the acute stages following TBI (Kuwar et al., 2019).
Conclusion
The NLRP3 inflammasome, is a multiprotein complex activated in response to brain injury in a time dependent manner. Accumulating evidence demonstrated that the beneficial effects of various non-specific inhibitors in attenuating inflammatory responses is via inhibition of NLRP3 inflammasome in TBI. These beneficial effects are mediated by modulating oxidative stress and the consequent TXNIP inhibition signify the potential of targeting TXNIP as a possible therapeutic option for the attenuation of NLRP3 inflammasome activation. However, it should be noted that none of the available antioxidants have proven efficacy in clinical trials. Moreover, there is no specific inhibitor for TXNIP.MCC950, specific small molecular NLRP3 inflammasome inhibitor, can cross the BBB and thereby by modulate inflammatory response. Although these studies have clinical implications, limited number of studies validated the beneficial effect of MCC950 in the setting of TBI. Further longterm functional investigation is required to suggest MCC950 as a promising new therapeutic candidate for patients with TBI.
Author contributions:SI wrote the manuscript; KP prepared the figures; TA prepared table, performed the literature survey; HA, TI, PT, SI contributed to critically reviewing the manuscript. All authors approved the final manuscript.
Conflicts of interest:The authors declare no conflicts of interest.
Financial support:This work was supported by the Department of Anatomy Neurobiology, UTHSC Memphis TN (to TI) and National Institute of Health, No. R01-NS097800 (to TI).
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-Non-Commercial-ShareAlike 4.0 License, which allows others to remix, tweak,and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
 中國(guó)神經(jīng)再生研究(英文版)2021年1期
中國(guó)神經(jīng)再生研究(英文版)2021年1期
- 中國(guó)神經(jīng)再生研究(英文版)的其它文章
- Oxidative stress battles neuronal Bcl-xL in a fight to the death
- Development and postnatal neurogenesis in the retina:a comparison between altricial and precocial bird species
- The role of peptidase neurolysin in neuroprotection and neural repair after stroke
- Cathepsins in neuronal plasticity
- Cognitive impairment in multiple sclerosis: lessons from cerebrospinal fluid biomarkers
- Progenies of NG2 glia: what do we learn from transgenic mouse models ?