
Normobaric oxygen therapy attenuates hyperglycolysis in ischemic stroke
2021-11-26 09:47ZheChengFengWuLiChristopherStoneKennethElkinChangYaPengRedinaBardhiXiaoKunGengYuChuanDing
中國(guó)神經(jīng)再生研究(英文版) 2021年6期
Zhe Cheng, Feng-Wu Li, Christopher R. Stone, Kenneth Elkin, Chang-Ya Peng,Redina Bardhi, Xiao-Kun Geng, , , , Yu-Chuan Ding, ,
Abstract Normobaric oxygen therapy has gained attention as a simple and convenient means of achieving neuroprotection against the pathogenic cascade initiated by acute ischemic stroke. The mechanisms underlying the neuroprotective efficacy of normobaric oxygen therapy, however,have not been fully elucidated. It is hypothesized that cerebral hyperglycolysis is involved in the neuroprotection of normobaric oxygen therapy against ischemic stroke. In this study, Sprague-Dawley rats were subjected to either 2-hour middle cerebral artery occlusion followed by 3- or 24-hour reperfusion or to a permanent middle cerebral artery occlusion event. At 2 hours after the onset of ischemia, all rats received either 95% oxygen normobaric oxygen therapy for 3 hours or room air. Compared with room air, normobaric oxygen therapy significantly reduced the infarct volume, neurological deficits, and reactive oxygen species and increased the production of adenosine triphosphate in ischemic rats. These changes were associated with reduced transcriptional and translational levels of the hyperglycolytic enzymes glucose transporter 1 and 3, phosphofructokinase 1, and lactate dehydrogenase. In addition, normobaric oxygen therapy significantly reduced adenosine monophosphate-activated protein kinase mRNA expression and phosphorylated adenosine monophosphate-activated protein kinase protein expression. These findings suggest that normobaric oxygen therapy can reduce hyperglycolysis through modulating the adenosine monophosphate-activated protein kinase signaling pathway and alleviating oxidative injury, thereby exhibiting neuroprotective effects in ischemic stroke. This study was approved by the Institutional Animal Investigation Committee of Capital Medical University (approval No. AEEI-2018-033) on August 13, 2018.
Key Words: neuroprotection; oxidative stress; oxygen; pathways; rat; recovery; repair; stroke Chinese Library Classification No. R459.6; R742; Q591.4
Introduction
Although stroke is both a leading cause of mortality and a major cause of serious long-term disability worldwide, the therapeutic options available to treat this deadly disease are currently limited (Chandra et al., 2017). Given the many potential complications and limitations of reperfusion therapy,however, the vast majority of acute ischemic stroke patients have not benefited from this strategy (Goyal et al., 2016).Clearly, additional adjunctive neuroprotective treatment strategies remain urgently needed.
Decreased cerebral blood flow and the tissue hypoxia it precipitates are crucial contributors to neurological deterioration after stroke (Singhal et al., 2005; Simon et al.,2019). Oxygen, which readily diffuses across the blood brain barrier, thus carries the potential to mitigate this deterioration by counteracting hypoxic brain injury. Normobaric oxygen(NBO), a non-invasive, inexpensive, familiar, and technically simple intervention requiring merely the availability of highflow oxygen and a facemask by which to deliver it, has emerged as a means of realizing this potential (Singhal et al., 2005). Indeed, rat models of acute ischemic stroke have shown that NBO therapy results in reductions of both infarct volumes and neurological deficits (Cai et al., 2017; Lan et al.,2018; Yang et al., 2019). Additionally, the neuroprotective effect of NBO was observed at any stage of experimental stroke (Beker et al., 2015; Ejaz et al., 2016; Tiwari et al.,2016). However, the optimal duration and dose of NBO remain unknown. A 2-8-hour NBO treatment demonstrated neuroprotective capacity after ischemia/reperfusion (Yuan et al., 2014), and a high NBO concentration was found to more efficiently alleviate infarct volume (Yuan et al., 2014; Yang et al., 2019). Despite this, previous clinical trials have failed to demonstrate a consistent neuroprotective effect of NBO against the effects of ischemic stroke (Mazdeh et al., 2015;Poli et al., 2017; Ding et al., 2018).
Many aspects of the mechanism underlying the neuroprotective effect of NBO in acute ischemic stroke remain unclear. One such aspect is its effect on glycometabolism.Previous studies from our group have demonstrated that NBO reduced brain damage by counteracting the post-stroke oxidative cascade (Cai et al., 2016a, 2017), but it is unknown whether NBO also attenuates the deleterious changes in glycometabolism associated with stroke. Hyperglycolysis is a manifestation of unbalanced cerebral glucose metabolism in stroke and is induced by diminished blood flow during ischemia. In response to hypoxia, expression of glucose transporter 1 and 3 (Glut1 and Glut3) is increased (Cai et al., 2016b), and phosphofructokinase 1 (PFK-1), the glycolytic rate-limiting enzyme, is upregulated, resulting in hyperglycolysis (Lenzen, 2014). Following this high glycolytic flux, the lactate dehydrogenase (LDH) concentration rises and is harmful to cell survival in the ischemic penumbra (Li et al., 2013). A recent study implicated the participation of adenosine monophosphate-activated protein kinase (AMPK)in this cascade, suggesting that its activation in the context of ischemia-reperfusion injury exacerbates hyperglycolysis (Geng et al., 2019a). In light of its role in ischemic injury and its consequent importance as a therapeutic target, we assessed post-ischemic hyperglycolysis as a potential locus of NBOmediated neuroprotection.
Materials and Methods
Animals
All experimental procedures in the study were performed in accordance with the National Institutes of Health (USA)Guidelines for the Care and Use of Animals and approved by the Institutional Animal Investigation Committee of Capital Medical University (approval No. AEEI-2018-033) on August 13, 2018. All experiments were designed and reported according to the Animal Research: Reporting ofIn VivoExperiments (ARRIVE) guidelines.
A total of 64 male Sprague-Dawley rats (specific-pathogenfree, 3 months old, 280-300 g, Vital River Laboratory Animal Technology Co., Ltd., Beijing, China, License No.911101147003408076) were randomly divided into four groups as follows: (1) sham-operated group without middle cerebral artery occlusion (MCAO) (n= 16), (2) 2 hours of MCAO + 3 hours of reperfusion (n= 16), (3) 2 hours of MCAO+ 24 hours of reperfusion (n= 16), and (4) permanent (28 hours) MCAO without reperfusion (pMCAO) (n= 16). Each group was randomly and equally divided into two subgroups that received either NBO or room air. All data were analyzed in a researcher-blinded manner.
Focal cerebral ischemia
The night before surgery, rats were fasted. Two hours of MCAO(right side) was achieved using the intraluminal filament technique (Kruyt et al., 2010); reperfusion was achieved after withdrawal of the filament. Physiological parameters,including the mean arterial pressure, blood partial pressure of carbon dioxide and partial pressure of oxygen, and rectal temperature, were monitored throughout the procedure.The rectal temperature was maintained at 36.5°C to 37.5°C through the use of heating lamps and pads. The mortality rate was less than 10% and was approximately equal in the NBO and room air subgroups. All deaths were caused by skull base hemorrhage because of arterial rupture during filament insertion or other operative procedures, rather than the ischemic time.
NBO treatment
NBO was delivered in a sealed chamber (50 cm × 25 cm × 25 cm) filled with 95% oxygen for 3 hours after 2 hours of MCAO.All rats in the four treatment groups received the same amount of NBO therapy. The oxygen concentration and flow rate (2 L/minute) were maintained using an oxygen controller(PRO-OX110; Reming Bioinstruments Co., Redfield, NY, USA).Carbon dioxide was continuously removed by soda lime(Sigma, Santa Clara, CA, USA) located at the bottom of the chamber.
Neurobehavioral functional scoring
All rats had normal neurological function prior to MCAO as assessed by the modified scoring system (which results in scores of 0-12) proposed by Beayev et al. (1996). Neurological deficits were assessed by this scoring system in rats after 24 hours of reperfusion or after 28 hours of pMCAO. Higher scores indicate more severe deficits.
Cerebral infarct volume
After 24 hours of reperfusion or 28 hours of pMCAO, brains were removed from ischemic rats, cut into 2-mm-thick slices(brain matrix), and stained with 2,3,5-triphenyltetrazolium chloride (Sigma). 2,3,5-Triphenyltetrazolium chloride staining has been widely used to quantify infarct volume in rats subjected to MCAO (Cipolla et al., 2017; Fan et al., 2018;Han et al., 2018). To minimize error caused by edema, the infarct volume was calculated using an indirect method by determining the volume as a percentage of the total ipsilateral brain volume (Wang et al., 2012).
Adenosine diphosphate/adenosine triphosphate ratio assay
After 3/24 hours of reperfusion or 28 hours of MCAO without reperfusion, ipsilateral ischemic cerebral hemispheres were dissected and processed. The adenosine diphosphate (ADP)/adenosine triphosphate (ATP) ratio assay was performed using the BioVision Assay Kit (Abcam, Cambridge, MA, USA)(Yuan et al., 2012). ATP and ADP levels were detected through luminescence by a DTX-880 Multimode Detector (Beckman Coulter, Bria, CA, USA). ADP/ATP ratios were subsequently calculated.
Reactive oxygen species production
The reactive oxygen species (ROS) assay was performed using a method that we have described previously (Geng et al.,2013). After 3/24 hours of reperfusion or 28 hours of pMCAO without reperfusion, brain samples taken from ipsilateral ischemic cerebral hemispheres were homogenized and then diluted to 10 mg/mL. After 30 minutes of incubation with 100 μg/mL digitonin, H2O2levels in brain homogenates were determined using 0.1 U/mL horseradish peroxidase, 50 μM Amplex red, and respiratory substrates [4 mM pyruvate, 2 mM malate, 0.8 mM oligomycin (complex V inhibitor), and 2 mM glutamate]. Fluorescence (emission 595 nm, excitation 535 nm) was then detected for 15 minutes at 37°C on a DTX-880 Multimode Detector (Beckman Coulter).
Reverse transcription polymerase chain reaction
A sensitive real-time reverse-transcription polymerase chain reaction technique was used in this study as described previously (Geng et al., 2015b). Briefly, total RNA from 100 mg of homogenized brain tissue samples was isolated using the Trizol reagent (Life Technologies, Carlsbad, CA, USA) and then converted into cDNA using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA).Quantification of gene expression was performed with a Prism 7500 real-time PCR system (Applied Biosystems). Expression of the target genes (Glut1, Glut3, LDH, PFK-1, and AMPK) from the experimental groups is represented as the fold-difference in expression relative to β-actin. The primer sequences were as follows: Glut1: forward: 5′-CGC CCT GTC CAG ACA CA-3′,reverse: 5′-CCT CCT AGC CAT TGT TCA GT-3′; Glut3: forward:5′-AAC ACC GGA GTC ATC AAT GC-3′, reverse: 5′-GCT GCG CTC TGT AGG ATA GC-3′; LDH: forward: 5’-CCG TTA CCT GAT GGG AGA AA-3′, reverse: 5′-AGC AGG GTA GAG CTG TGG AA-3′;PFK-1: forward: 5′-ATT GAA TAT GCC GTC TCC-3′, reverse: 5′-CAC AAG ATA CAC ACA TGG-3′; AMPK: forward: 5′-TGT GAC AAG CAC ATT TTC CAA-3′, reverse: 5′-CCG ATC TCT GTG GAG TAG CAG-3′.
Western blot assay
A western blot assay was used to quantify protein expression at 3 and 24 hours after reperfusion, as well as after 28 hours of pMCAO. Proteins extracted from ipsilateral ischemic cerebral hemispheres including the middle cerebral arterysupplied territory (cortex and striatum) were loaded onto sodium dodecyl sulphate-polyacrylamide gels for electrophoresis. Upon conclusion of electrophoresis, proteins were transferred to a polyvinylidene difluoride membrane and incubated with primary antibodies [rabbit anti-Glut1(1:400; Cat# Sc-7903; Santa Cruz Biotechnology, Santa Cruz,CA, USA), rabbit anti-Glut3 (1:400; Cat# Sc-31838; Santa Cruz Biotechnology), rabbit anti-PFK-1 (1:5000; Cat# 200-1156-0100; Rockland Immunochemicals, Inc., Gilbert, CA, USA),rabbit anti-LDH (1:2000; Cat# 200-1173-0100; Rockland Immunochemicals), rabbit anti-AMPK (1:500; Cat# Sc-19128;Santa Cruz Biotechnology, Inc.), and rabbit anti-pAMPK (1:500;Cat# Sc-101630; Santa Cruz Biotechnology)] for 24 hours at 4°C. The secondary antibody used for all primary antibodies was goat anti-rabbit IgG-horseradish peroxidase (Cat# sc-2004;Santa Cruz Biotechnology), and incubation was performed for 1 hour at room temperature. To quantify protein expression,western blot images were analyzed in terms of relative image density using ImageJ 1.42 software (National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
Statistical analysis was performed using SPSS version 17(IBM, Armonk, NY, USA). Differences among multiple groups were assessed using one-way analysis of variance.Post hoccomparisons between groups were performed using the least significant difference test. The significance level was set atP<0.05.
Results
Effect of NBO on the physiological parameters in ischemic stroke rats
There were no significant differences in the mean arterial pressure, blood glucose, blood pH, or partial pressure of carbon dioxide among the pMCAO, 2-hour MCAO, and 2-hour MCAO + NBO groups (P> 0.05). NBO treatment significantly elevated the partial pressure of oxygen in MCAO rats compared with room air (P< 0.01; Table 1). Body temperature(rectal site) was maintained at approximately 37°C.
NBO improves the infarct volume and neurological deficits in ischemic stroke rats
At 24 hours after reperfusion, NBO produced a significant decrease in infarct volume compared with 2 hours of MCAO with room air (P< 0.05; Figure 1A and C); in contrast, NBO did not induce significant neuroprotection in the pMCAO group. Neurobehavioral testing performed at 24 hours after reperfusion demonstrated significantly reduced neurological deficits in the MCAO + NBO group compared with room air (P< 0.05; Figure 1B). NBO did not significantly reduce
neurological deficits in the pMCAO group.
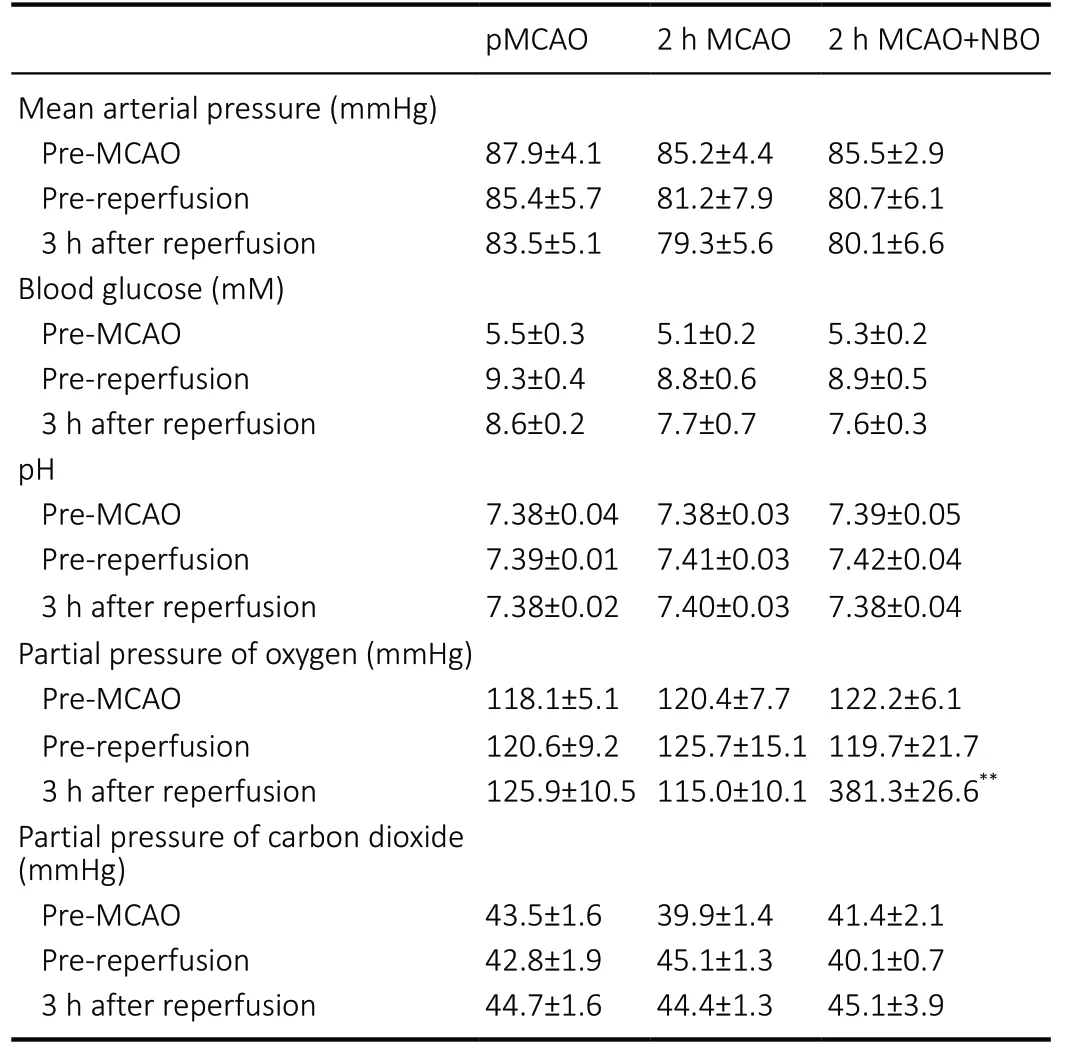
Table 1 |Effect of NBO on the physiological parameters of ischemic stroke rats
NBO increases the energy metabolism in cerebral tissue of ischemic stroke rats
The metabolic viability of cerebral tissue was assessed by measuring the ADP/ATP ratio. Compared with the shamoperated group (referenced as 1, data not shown), the ADP/ATP ratio was significantly elevated at 3 (P< 0.01) and 24 hours (P< 0.01) after reperfusion, as well as after pMCAO (P< 0.01),indicating that an impairment of energy metabolism was induced. Compared with room air, NBO significantly decreased the ADP/ATP ratio at 3 (P< 0.01) and 24 hours (P< 0.01) after reperfusion, indicating that ATP levels were preserved by NBO after ischemia/reperfusion. NBO also decreased the ADP/ATP ratio in the pMCAO group (P< 0.01,vs. room air; Figure 2A).
NBO increases the oxidative stress in cerebral tissue of ischemic stroke ratsROS levels were significantly increased at 3 hours (P<0.01) and remained elevated at 24 hours (P< 0.01) after reperfusion, as well as after pMCAO (P< 0.01), compared with the sham-operated group (referenced as 1, data not shown).NBO treatment significantly decreased ROS levels at 3 (P<0.05) and 24 hours (P< 0.05) after reperfusion compared with the room air groups. NBO therapy did not yield a significant effect on ROS levels in the pMCAO group (Figure 2B).
NBO decreases glycolysis in cerebral tissue of ischemic stroke rats
The mRNA and protein expression levels of the two major glucose transporters, Glut1 and Glut3, were measured to evaluate the state of glycolysis in the context of NBO treatment after ischemia/reperfusion. Compared with the sham-operated group (referenced as 1, data not shown),mRNA and protein expression of Glut1 were significantly increased at 3 (P< 0.01) and 24 hours (P< 0.01) after reperfusion and after pMCAO (P< 0.01). Compared with room air, Glut1 mRNA expression was significantly decreased at 3 (P< 0.01) and 24 hours (P< 0.01) after reperfusion by NBO; this effect was not observed in the pMCAO group (Figure 3A). NBO significantly decreased protein expression of Glut1 at 3 (P<0.01) and 24 hours (P< 0.01) after reperfusion. In the pMCAO group, protein expression of Glut1 was also decreased after NBO therapy (P< 0.01,vs. room air; Figure 3B).
Compared with the sham-operated group (referenced as 1, data not shown), mRNA and protein expression of Glut3 was significantly increased at 3 (P< 0.01) and 24 hours after reperfusion (P< 0.01), as was mRNA expression of Glut3 in the pMCAO group (P< 0.01). NBO decreased Glut3 mRNA expression in all groups, although not significantly (Figure 3C). Decreases induced by NBO in Glut3 protein expression at 3 (P< 0.01) and 24 hours (P< 0.01) after reperfusion were significant. NBO therapy did not yield a significant effect on Glut3 protein expression in the pMCAO group (Figure 3D).
NBO inhibits hyperglycolysis in cerebral tissue of ischemic stroke rats
The expression levels of PFK-1 and LDH were assessed because of the importance of these enzymes to the hyperglycolytic process (Lenzen, 2014; Melkonian and Schury, 2020).Compared with the sham-operated group (referenced as 1,data not shown), mRNA expression of PFK-1 was significantly increased at 24 hours after reperfusion (P< 0.01), and protein expression was increased at 3 (P< 0.05) and 24 hours (P<0.01) after reperfusion; protein expression of PFK-1 was also increased in the pMCAO group (P< 0.01). NBO treatment significantly reduced PFK-1 mRNA expression at both 3 and 24 hours after reperfusion (bothP< 0.01) but not in the pMCAO group. NBO treatment also significantly decreased protein expression of PFK-1 at 3 (P< 0.01) and 24 hours (P<0.01) after reperfusion and in the pMCAO group (P< 0.01),compared with room air (Figure 4A and B).
In addition, compared with the sham-operated group(referenced as 1, data not shown), LDH mRNA expression was significantly increased at 24 hours after reperfusion (P< 0.01).Significant increases in LDH protein expression were observed at 3 (P< 0.05) and 24 hours (P< 0.01) after reperfusion and in the pMCAO group (P< 0.05). In the NBO group, LDH mRNA expression was significantly reduced at both 3 (P< 0.01) and 24 hours (P< 0.01) after reperfusion, while no significant effect on LDH mRNA expression was observed in the pMCAO group (Figure 4C). With NBO treatment, LDH protein expression was reduced in the MCAO groups at 3 (P< 0.01)and 24 hours (P< 0.01) after reperfusion, as well as in the pMCAO group (P< 0.05; Figure 4D).
NBO inhibits AMPK signaling in cerebral tissue of ischemic stroke rats
Compared with the sham-operated group (referenced as 1,data not shown), mRNA expression of AMPK was significantly increased at 3 and 24 hours after reperfusion (bothP< 0.01).pAMPK protein expression was also increased significantly at 3 and 24 hours after reperfusion (P< 0.01). NBO significantly reduced AMPK mRNA expression at 3 (P< 0.01) and 24 hours(P< 0.01) after reperfusion (Figure 5A), did not produce an effect on AMPK protein expression (Figure 5B), and significantly reduced pAMPK protein expression at 3 (P< 0.01)and 24 hours (P< 0.05) after reperfusion (Figure 5C). Although pAMPK protein expression was slightly increased in the pMCAO group, no significant effect of NBO was demonstrated in this group.
Discussion
In this study, we demonstrate that NBO significantly decreased infarct volume, neurological deficits, and ROS and also increased ATP production. Furthermore, the reduced oxidative damage and improved energy metabolism in the brain were accompanied by decreased expression of the molecular markers of hyperglycolysis, including Glut1, Glut3,PFK-1, and LDH, at both the transcriptional and translational levels. In addition, NBO significantly reduced the mRNA and protein expression of pAMPK, both of which were upregulated by ischemia, thus implicating AMPK signaling in the hyperglycolytic cascade. These findings suggest that NBOmediated neuroprotection may proceed through attenuation of hyperglycolysis after ischemic stroke.
This neuroprotective efficacy of NBO therapy has been demonstrated in numerous studies after ischemic stroke in rats(Lan et al., 2018) and has manifested as mitigation of neuronal death and extension of the time window for reperfusion(Kim et al., 2005; You et al., 2016). To date, multiple physiological changes have been proposed to account for this neuroprotection, including increased tissue oxygenation,increased cerebral blood flow, reduced oxidative stress, and protection of blood-brain barrier integrity (Qi et al., 2013).Studies performed previously by our group demonstrated that NBO treatment significantly augments cell survival in rats compared with alteplase alone by downregulating nicotinamide adenine dinucleotide phosphate oxidase activity and subunit expression (Geng et al., 2013; Ji et al., 2015).In addition, our data indicated that NBO may yield superior effects when used in conjunction with ethanol through the combined improvements in cellular metabolism produced by this regimen (Geng et al., 2015a, b; Cai et al., 2016a, 2017).Recently, a randomized control trial conducted by our group provided another potentially efficacious combination therapy incorporating NBO by suggesting that NBO might improve the functional prognosis in arterial ischemic stroke patients after mechanical thrombectomy (Geng et al., 2019b).
The encouraging results of these individual studies notwithstanding, the overall inconsistent clinical outcomes with which NBO therapy research has been afflicted demand further exploration of the mechanisms underlying NBOassociated effects. Oxidative stress, or the overproduction of ROS, is a fundamental damage mechanism after stroke that results in increased brain edema, hemorrhagic transformation,cell death, and infarct volume (Li and Yang, 2016). The shift to glycolysis during ischemia and the subsequent acidosis enhance ROS production (Kochanski et al., 2013); thus, in addition to its impact on cellular energetics, glucose handling in the ischemic brain is also intimately linked to cellular damage mechanisms. Given this link, demonstration of the capacity of NBO to modulate hyperglycolysis and the AMPK pathway after stroke would not only elucidate the basis of NBO-mediated neuroprotection, but also reveal a pathway for which adjuvant therapies may be developed to enhance the beneficial effect of NBO. In this study, we contributed to this demonstration by investigating the impact of NBO on hyperglycolysis in the MCAO rat model and finding a significant, NBO-mediated decrease in hyperglycolysis in the ischemic brain following reperfusion.
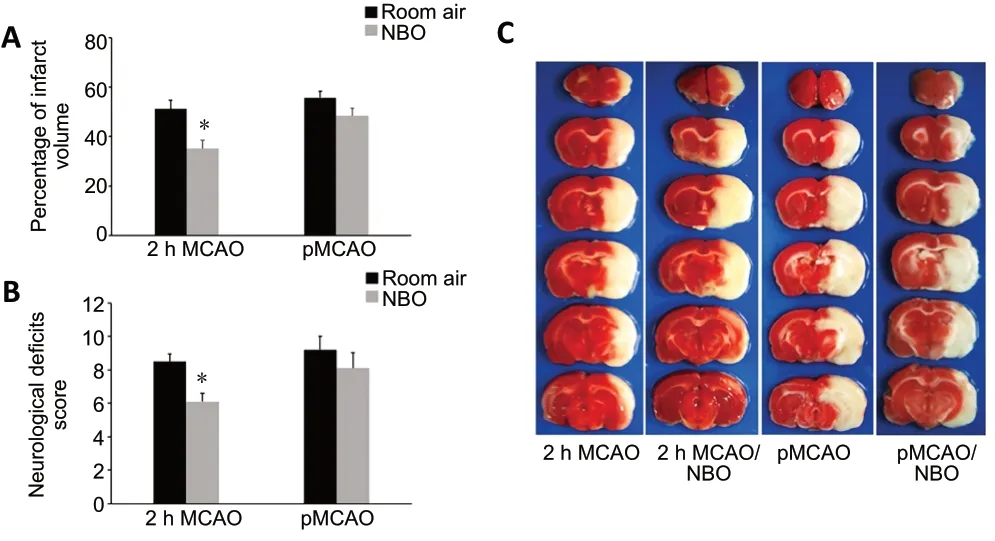
Figure 1 |Effect of NBO on the infarct volume and neurological deficits in ischemic stroke rats.
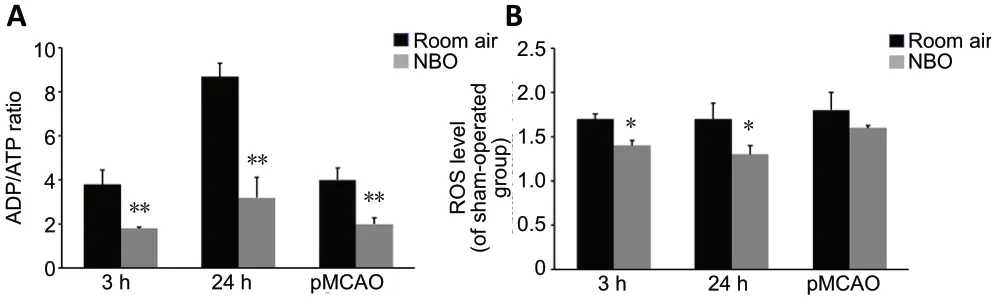
Figure 2 |Effect of NBO on the ADP/ATP ratio (A) and ROS levels (B) in cerebral tissue of ischemic stroke rats.
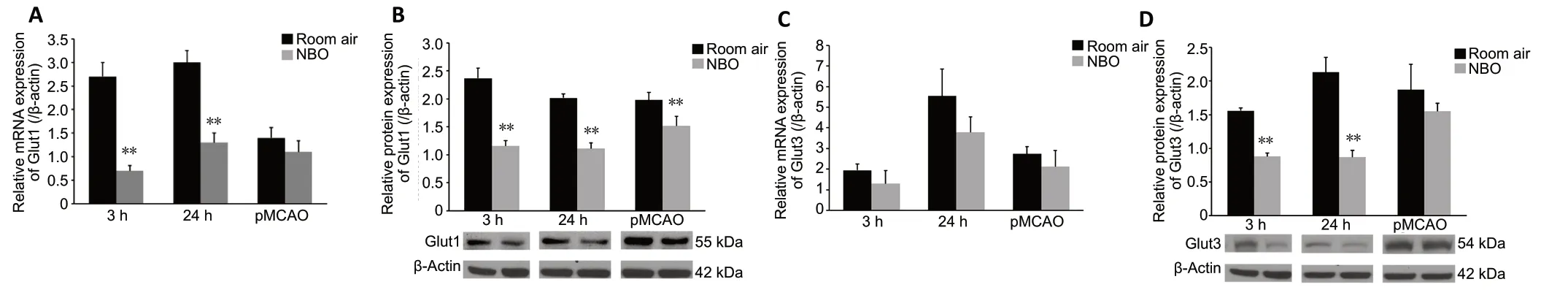
Figure 3 |Effect of NBO on Glut1 and Glut3 expression in cerebral tissue of ischemic stroke rats.
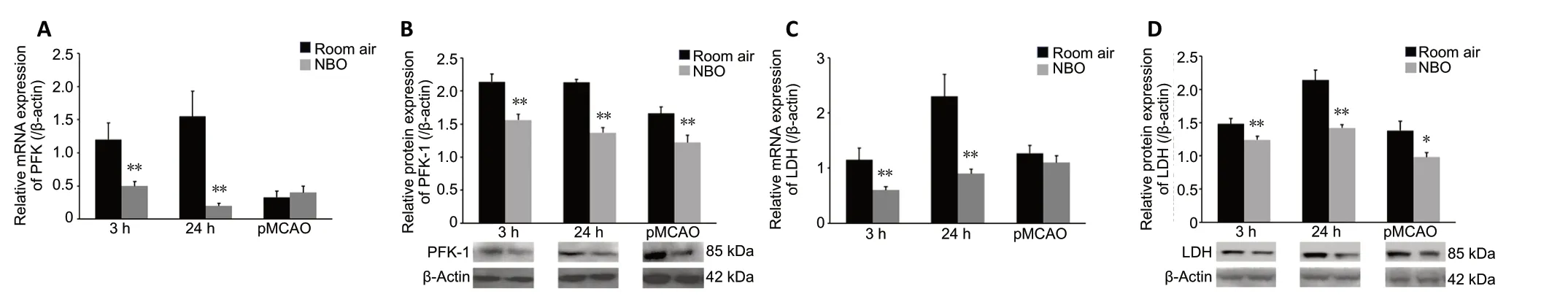
Figure 4 |Effect of NBO on PFK-1 and LDH expression in cerebral tissue of ischemic stroke rats.
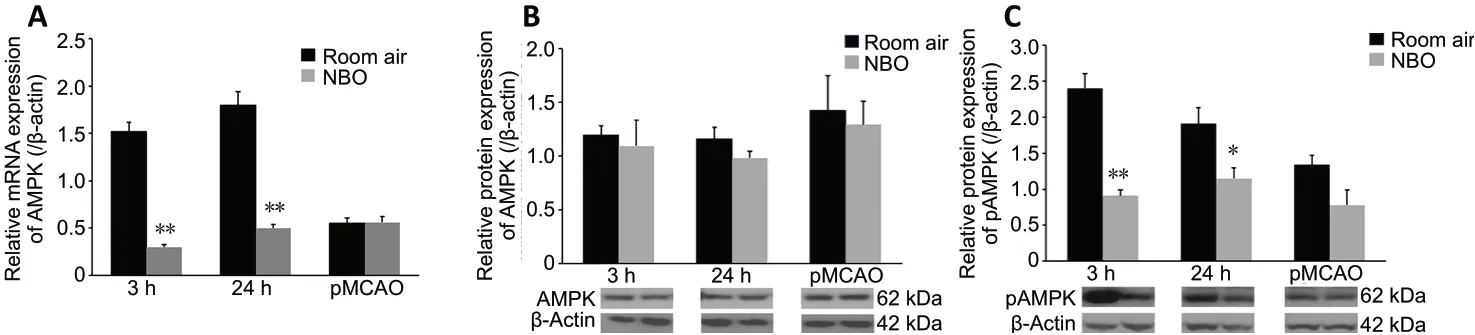
Figure 5 |Effect of NBO on AMPK expression in cerebral tissue of ischemic stroke rats.
Two major metabolic pathways by which ATP is generated in mammalian cells include oxidative phosphorylation in the mitochondria and glycolysis in the cytosol. Hypoxia can induce mitochondrial dysfunction in many cells (Qiu et al.,2016; Laouafa et al., 2019; Liu et al., 2020), and mitochondrial dysfunction has been suggested to upregulate glycolysis to compensate for the consequent loss of energy generation by oxidative phosphorylation (Hu et al., 2012). Hyperglycolysis is characterized by increased uptake and utilization of glucose relative to oxygen consumption (Cai et al., 2016b). This has been hypothesized to represent a defensive response by neural cells designed to alleviate the extreme depression of brain glucose levels seen under ischemic conditions (Li et al., 2018). Despite the ostensibly beneficial consequence of increased ATP production in neurons, the sudden elevation of oxygen-deprived metabolism can lead to the buildup of lactate(Rogatzki et al., 2015). Thus, the emergence of hyperglycolysis after ischemia-reperfusion promotes a toxic accumulation of lactic acid and ROS (Shen et al., 2019). This is attributable to the tendency in hypoxic cells toward an elevation of anaerobic glycolysis over oxidative metabolism, during which the change in redox state precipitated by hypoxia promotes the reduction of pyruvate to lactate by LDH (Rogatzki et al.,2015). Conversion of NADH to NAD+by LDH in this reaction sustains glycolysis (Melkonian and Schury, 2020), but the resultant accumulation of lactic acid reduces the intracellular pH, initiating or enhancing apoptotic cell death in the ischemic penumbra (Uzdensky, 2020). The finding in our study that NBO decreased LDH expression suggests that this therapy may interrupt hyperglycolysis and, therefore, may reduce acidosis and apoptosis after ischemia-reperfusion. The reduction in ROS generation observed in our study provides further support for the prospect of an NBO-mediated reduction in apoptosis (Franklin, 2011).
Previous work has linked hyperglycolysis to poor functional outcome and high mortality in patients with hyperglycemia(Marulaiah et al., 2017). Compared with oxidative phosphorylation, hyperglycolysis is inefficient and involves upregulation of the Glut1 and Glut3 proteins to enhance glycolysis in the context of ischemia (Vannucci et al.,1996; Zhang et al., 2009; Xia et al., 2018). In this study,the reductions in Glut1 and Glut3 expression seen in the experimental groups suggest that NBO therapy may inhibit the uptake and utilization of glucose, potentially counteracting the effects of post-stroke hyperglycolysis. PFK-1, a key ratelimiting enzyme in glycolysis (Lenzen, 2014), is activated after stroke by hypoxia and by the increase in ADP (Dornbos et al., 2013). The capacity of NBO to reduce PFK-1 expression,as demonstrated by our results, further suggests that NBO therapy may attenuate hyperglycolysis.
The decreased expression of LDH, Glut1, Glut3, and PFK-1 after NBO treatment strongly indicates a mechanism involving blunted glucose uptake and utilization in hyperglycolysis. A consequence of this decrease in hyperglycolytic imbalance suggested in our study by the reduced ADP/ATP ratio is that NBO may improve overall neuronal energy metabolism. The results of our biochemical analyses implicate the involvement of AMPK in these changes. AMPK has been reported to be an important regulatory component of glycometabolism that serves to match energy supply with energy demand (Barnes et al., 2002). Suppressions of energy supply (e.g., in poststroke hypoxia) activate AMPK via an increase in the AMP/ATP ratio. This increase then promotes ATP generation,potentially through increased glucose uptake and glycolysis(Marsin et al., 2002). Evidence of a connection between AMPK and glycolytic enzymes has been produced in the context of cardiac ischemia, in which AMPK appears to increase glycolysis through phosphorylation and activation of PFK-2 (Marsin et al., 2000). PFK-2 catalyzes the production of fructose 2,6-bisphosphate, which then stimulates PFK-1 (Shen et al., 2006), a key rate-limiting enzyme in glycolysis (Lenzen,2014). Although AMPK regulation is an adaptive response to ischemic conditions and plays a vital role in maintaining energy homeostasis, the overactivation of AMPK after stroke can aggravate hyperglycolysis (Marsin et al., 2002), leading to maladaptive metabolic consequences. In our study, NBO treatment was followed by a reduction in pAMPK in concert with decreased expression of Glut1, Glut3, and PFK-1,indicating that AMPK may be downregulated by NBO-activated metabolic signals.
Some limitations should be considered when evaluating the significance of the results obtained in this study. First, in contrast to other work that assessed behavioral consequences several weeks after MCAO (Tatarishvili et al., 2014), the behavioral outcome was assessed in our study at 24-48 hours post-stroke. Because the neuroprotective mechanism by which NBO therapy appears to operate alleviates acute phase injury, this study was designed to focus on immediate outcomes. In future work, we will expand on our current findings by assessing long-term functional outcomes. Another limitation involves the young age of the rats utilized in this experiment; in contrast to other ischemic stroke studies in which aged rats were used (Badan et al., 2003; Popa-Wagner et al., 2006; Joseph et al., 2012), young rats may exhibit better recovery and may benefit from more effective NBO-mediated attenuation of hyperglycolysis. Thus, a future study will be designed using aged rats to account for this variable. Finally,the effects of a variety of NBO doses should be explored,both in the interest of designing an optimal regimen for translational research and to evaluate the possible existence of dose thresholds above which complications may arise.
In conclusion, this study demonstrates that a short duration of NBO therapy delivered in the wake of ischemia-reperfusion injury produces a neuroprotective effect through a reduction of hyperglycolysis in association with reductions in AMPK mRNA expression and pAMPK protein expression, both of which were upregulated by ischemia. This finding provides additional insight into the efficacy of NBO treatment and may assist in the design of optimal therapeutic NBO regimens for post-stroke patients.
Author contributions:Study conception and design: ZC, XKG, YCD; animal and biochemical experiment and data analysis: ZC, CYP; manuscript writing: ZC, FWL; manuscript revision: CRS, KE, RB, XKG, YCD. All authors approved the final version of the manuscript.
Conflicts of interest:None declared.
Financial support:This study was partially supported by the National Natural Science Foundation of China, Nos. 81802231 (to FWL), 81871838(to XKG), the Organization Department of Beijing Talents Project, No.2018000082595G485 (to FWL), and the Science and Technology Plan of Beijing Tongzhou District of China, No. KJ2019CX004 (to ZC and FWL).The funder had no roles in the study design, conduction of experiment,data collection and analysis, decision to publish, or preparation of the manuscript.
Institutional review board statement:The study was approved by the Institutional Animal Investigation Committee of Capital Medical University(approval No. AEEI-2018-033) on August 13, 2018.
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement:Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix,tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewers:Aurel Popa-Wagner, University Medicine Rostock,Germany; Lorenzo Romero-Ramíre, Unidad de Neurologia Experimental,Hospital Nacional de Paraplejicos (SESCAM), Spain; Julien Rossignol, Central Michigan University, USA; Eric Schmidlin, University of Fribourg Medicine,Switzerland.
Additional files:
Additional file 1:Open peer review reports 1-4.
Additional file 2:Original data of the experiment.
 中國(guó)神經(jīng)再生研究(英文版)2021年6期
中國(guó)神經(jīng)再生研究(英文版)2021年6期
- 中國(guó)神經(jīng)再生研究(英文版)的其它文章
- Entacapone promotes hippocampal neurogenesis in mice
- Electroacupuncture improves learning and memory functions in a rat cerebral ischemia/reperfusion injury model through PI3K/Akt signaling pathway activation
- MicroRNA-670 aggravates cerebral ischemia/reperfusion injury via the Yap pathway
- Corticospinal excitability during motor imagery is diminished by continuous repetition-induced fatigue
- TP53-induced glycolysis and apoptosis regulator alleviates hypoxia/ischemia-induced microglial pyroptosis and ischemic brain damage
- Apelin-13 inhibits apoptosis and excessive autophagy in cerebral ischemia/reperfusion injury