
TP53-induced glycolysis and apoptosis regulator alleviates hypoxia/ischemia-induced microglial pyroptosis and ischemic brain damage
2021-11-26 09:48LanLanTanXiaoLuJiangLiXiaoXuGenLiChenXiFengXinDingBinSunZhengHongQinZuBinZhangXingFengMeiLi
中國神經(jīng)再生研究(英文版) 2021年6期
Lan-Lan Tan , Xiao-Lu Jiang , Li-Xiao Xu, Gen Li, Chen-Xi Feng, Xin DingBin Sun Zheng-Hong Qin, Zu-Bin Zhang, , Xing Feng , Mei Li,
Abstract Our previous studies have demonstrated that TP53-induced glycolysis and apoptosis regulator (TIGAR) can protect neurons after cerebral ischemia/reperfusion. However, the role of TIGAR in neonatal hypoxic-ischemic brain damage (HIBD) remains unknown. In the present study,7-day-old Sprague-Dawley rat models of HIBD were established by permanent occlusion of the left common carotid artery followed by 2-hour hypoxia. At 6 days before induction of HIBD, a lentiviral vector containing short hairpin RNA of either TIGAR or gasdermin D (LV-sh_TIGAR or LV-sh_GSDMD) was injected into the left lateral ventricle and striatum. Highly aggressively proliferating immortalized (HAPI) microglial cell models of in vitro HIBD were established by 2-hour oxygen/glucose deprivation followed by 24-hour reoxygenation. Three days before in vitro HIBD induction, HAPI microglial cells were transfected with LV-sh_TIGAR or LV-sh_GSDMD. Our results showed that TIGAR expression was increased in the neonatal rat cortex after HIBD and in HAPI microglial cells after oxygen/glucose deprivation/reoxygenation. Lentivirusmediated TIGAR knockdown in rats markedly worsened pyroptosis and brain damage after hypoxia/ischemia in vivo and in vitro. Application of exogenous nicotinamide adenine dinucleotide phosphate (NADPH) increased the NADPH level and the glutathione/oxidized glutathione ratio and decreased reactive oxygen species levels in HAPI microglial cells after oxygen/glucose deprivation/reoxygenation. Additionally, exogenous NADPH blocked the effects of TIGAR knockdown in neonatal HIBD in vivo and in vitro. These findings show that TIGAR can inhibit microglial pyroptosis and play a protective role in neonatal HIBD. The study was approved by the Animal Ethics Committee of Soochow University of China (approval No. 2017LW003) in 2017.
Key Words: hypoxic-ischemic brain damage; in vitro; in vivo; microglia; NADPH; pyroptosis; ROS; TIGAR Chinese Library Classification No. R453; R741; Q255
Introduction
Hypoxic-ischemic brain damage (HIBD) constitutes one of the major causes of high neonatal morbidity and neurological sequelae in fetuses and newborn infants worldwide(Psychogios et al., 2017; Jiang et al., 2020). Therapeutic hypothermia is widely used and is the gold standard of neonatal HIBD treatment (Chalak and Kaiser, 2007; Davies et al., 2019). However, this therapy has the shortcomings of a narrow therapeutic window and incomplete neuroprotection(Liu et al., 2017). Thus, further study is required to elucidate the underlying mechanism of HIBD and ultimately seek novel and potential therapeutic targets and approaches for HIBD treatment.
Pyroptosis, which is a novel type of proinflammatory programmed cell death, plays an important role in the pathological process of neurological diseases, such as Alzheimer’s disease and ischemic stroke (Ito et al., 2015;Barrington et al., 2017; Yagami et al., 2019). Pyroptosis is induced by the proteolytic activation of caspase-1 through cleaving the gasdermin D (GSDMD) N-terminal (GSDMD-N)(Kepp et al., 2010; Liu et al., 2018). GSDMD-N interacts with intimal lipids to form pores on the plasma membrane, which causes the cell membrane to rupture and release the proinflammatory mediator interleukin-1β (IL-1β) and ultimately leads to cell death (Sborgi et al., 2016; McKenzie et al., 2018;Xu et al., 2019b). Microglia, the resident innate immune cells in the central nervous system, are associated with the inflammatory response (Xu et al., 2019a; Xie et al., 2021).Microglial activation is considered to contribute to triggering neuroinflammation, reactive oxygen species (ROS) production,and cerebral injury via the release of pro-inflammatory cytokines (Arioz et al., 2019; Mo et al., 2020). Recent studies have shown that inflammasome-mediated microglial pyroptosis contributes to brain injury in neurological diseases including ischemic stroke, Parkinson’s disease, and traumatic brain injury(Sarkar et al., 2017; Lee et al., 2019; Poh et al., 2019). Inhibition of inositol-requiring enzyme 1α alleviates neuronal pyroptosis through the miR-125/NLR family pyrin domain containing 1 pathway and reduces neonatal rat hypoxic-ischemic brain injury (Huang et al., 2020). Recently, we found that inhibition of NLR family pyrin domain containing 3 (NLRP-3)/caspase-1/GSDMD axis-mediated microglial pyroptosis decreased brain injury in neonatal rats after HIBD (Lv et al., 2020), which implies that microglial pyroptosis plays a crucial role in neonatal HIBD.However, the underlying mechanisms that mediate microglial pyroptosis remain largely unclear.
TP53-induced glycolysis and apoptosis regulator (TIGAR)decreases fructose-2,6-bisphosphate levels in cells,subsequently leading to inhibition of glycolysis and a decrease in intracellular ROS levels (Bensaad et al., 2006). We previously found that TIGAR played a neuroprotective role in ischemic stroke by elevating the flux of the pentose phosphate pathway(PPP) (Li et al., 2014). Recently, we further demonstrated that TIGAR played a passive role in the astrocyte-mediated inflammatory response after ischemic stroke (Chen et al.,2018). Therefore, we hypothesized that TIGAR suppressed microglial pyroptosis and protected against neonatal HIBD by decreasing ROS. The present study tested this hypothesis in a neonatal rat hypoxic-ischemic brain injury modelin vivoand in highly aggressively proliferating immortalized (HAPI) microglial cellsin vitro.
Materials and Methods
Neonatal HIBD model
All animal experiments were conducted according to the institutional guidelines for animal care and use. Neonatal rats were freely fed by their mothers. The room temperature during feeding was 24 ± 2°C. The rats were maintained on a 12-hour light/dark cycle. The study protocol was approved by the Animal Ethics Committee of Soochow University of China(approval No. 2017LW003) in 2017. One hundred and fiftythree neonatal Sprague-Dawley rats (12-17 g, 7 days old)were purchased from Zhaoyan (Suzhou) New Drug Research Center Co., Ltd. (Suzhou, China, License No. SCXK (Su) 2018-0006). Neonatal rats were randomly divided into sham and HIBD groups. After viral infection, the HIBD group was divided into normal control, model, LV-sh_TIGAR, LV-sh_GSDMD,NADPH, and LV-sh_TIGAR + NADPH groups. The HIBD model was established according to a modified Vannucci method as described previously (Ding et al., 2015; Patel et al., 2015).Briefly, the left common carotid artery was permanently ligated in rats. After 1 hour of recovery with their mothers,the rats were subjected to hypoxia (8% O2/92% N2) at 37°C for 2 hours and then returned to their mothers for free feeding.Sham-operated rats underwent the same surgery but were not ligated or subjected to hypoxia. Neonatal rats were euthanized at the indicated time point after hypoxic-ischemic brain injury.
Oxygen and glucose deprivation/reoxygenation
HAPI rat microglial cell lines were purchased from the Cell Bank of Enzyme-Linked Biotechnology Co., Ltd. (Shanghai,China). HAPI microglial cells were cultured in Dulbecco’s modified Eagle medium (Gibco, Waltham, MA, USA)supplemented with 10% fetal bovine serum (Biological Industries, Kibbutz Beit-Haemek, Israel), 100 U/mL penicillin and 100 mg/mL streptomycin in a humidified atmosphere at 37°C with 5% CO2. The HAPI microglial cells were incubated with glucose-free Earle’s balanced salt solution and then placed in a chamber (ELECTROTEK, West Yorkshire, UK) with 95% N2and 5% CO2at 37°C for 2 hours. Control cells were incubated with glucose-free Earle’s balanced salt solution and then placed in normal culture conditions for 2 hours.Two hours after oxygen and glucose deprivation (OGD),the medium was replaced with normal culture medium(reoxygenation), and the cells were cultured at 37°C with 5%CO2for 24 hours.
Viral infection and drug treatment in vitro and in vivo
For viral infectionin vivo, 6 days prior to HIBD, a vector containing TIGAR short hairpin RNA (LV-sh_TIGAR; 1×109transduction units/mL; Genechem, Shanghai, China)or GSDMD short hairpin RNA (LV-sh_GSDMD; 1 × 109transduction U/mL; Genechem) was infused into the left lateral ventricle and striatum (2 μL/injection site) (Li et al.,2014) of the neonatal rats. Nicotinamide adenine dinucleotide phosphate (NADPH; 2.5 mg/kg; Beyotime, Nantong, China)was injected into the left lateral ventricle of the rat brain before HIBD modeling.
For viral infectionin vitro, the HAPI rat microglial cells were treated with medium containing LV-sh_TIGAR (MOI = 100), LVsh_GSDMD (MOI = 100), or LV-sh-scramble (normal control;MOI = 100; Genechem), and the medium was replaced with regular medium after 12 hours of viral treatment. The cells were cultured for 3 days and then subjected to OGD/reoxygenation (OGD/R). NADPH (10 μM; Beyotime) was added before OGD and was present in the regular medium used for reoxygenation at 24 hours.
In vitro cell death assay
Cell death was detected using a Cell Counting Kit-8 (Dojindo,Kumamoto, Japan) and a lactate dehydrogenase cytotoxicity assay kit (Nanjing Jiancheng Bioengineering Institute, Nanjing,China) according to the manufacturer’s protocols.
Measurement of NADPH, glutathione, and ROS levels
The levels of NADPH and glutathione (GSH) in cultured HAPI microglial cells were measured with an NADPH/NADP+kit(Beyotime) and a GSH/oxidized GSH (GSSG) kit (BioAssay Systems, San Francisco, CA, USA) following the manufacturer’s instructions. The ROS levels in HAPI microglial cells were detected using dihydroethidium staining (1 μM; Sigma, St.Louis, MO, USA) as previously described (Cao et al., 2017).
Western blot assay
Neonatal rats were deeply anesthetized with anhydrous 2-butanol and then sacrificed. Cortical tissues of the neonatal rat brain and HAPI microglial cells were lysed into homogenate in lysis buffer, and the extracted proteins (20 μg) were separated on 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels and transferred to nitrocellulose membranes (Millipore, Burlington, MA, USA). Membranes were incubated with mouse anti-TIGAR (1:1000; Cat# sc-166290; Santa Cruz Biotechnology, Santa Cruz, CA, USA), rabbit anti-Caspase-1 (1:1000; Cat# ab207802; Abcam, Cambridge,UK), rabbit anti-GSDMD (1:1000; Cat# ab219800; Abcam),anti-GSDMD-N (1:1000; Cat# ab215203; Abcam), rabbit anti-IL-1β (1:1000; Cat# ab9787; Abcam), or mouse anti-β-actin(1:10 000; Cat# ab008; MULTI SCIENCES, Hangzhou, China)overnight at 4°C. The membranes were then incubated with peroxidase-conjugated rabbit/mouse secondary antibodies(1:5000; Cat# 111-035-003/115-035-003; Jackson, Lancaster,PA, USA), and bands were detected with an AmerSham Imager 600 (GE, Boston, MA, USA) and analyzed using ImageJ analysis software (National Institutes of Health, Bethesda, MD, USA).
2,3,5-Triphenyltetrazolium chloride staining
Cerebral lesion infarctions were measured 24 hours after HIBD by 2,3,5-triphenyltetrazolium chloride staining (Sinopharm Chemical Reagent Co., Ltd., Shanghai, China) as described previously (Cao et al., 2017). The rats were sacrificed under deep anesthesia with anhydrous 2-butanol. The brain was removed immediately and sliced into parallel 2-mm-thick slices, immersed in 2,3,5-triphenyltetrazolium chloride solution for 5 minutes, and fixed in 4% paraformaldehyde solution. The cerebral infarction lesion was measured as a percentage of the total hemisphere.
Hematoxylin and eosin staining and immunohistochemistry
Twenty-four hours after HIBD, the rat brains were removed and soaked in 20% and 30% sucrose for gradient dehydration.Brain sections (10 μm) were cut with a paraffin slicer (SLEE,Mainz, Germany) and then stained with hematoxylin and eosin. Briefly, the brain sections were immersed in xylene and alcohol and were then stained with hematoxylin. After washing with tap water, the sections were stained with eosin and sealed with neutral gum.
Brain sections (4 μm) were subjected to immunohistochemistry.The brain sections were immersed in xylene, anhydrous ethanol, and alcohol. The sections were then incubated with rabbit anti-GSDMD-N antibody (1:500; Cell Signaling Technology, Inc., Beverly, MA, USA) overnight at 4°C and were next incubated with HRP-labeled goat anti-rabbit IgG(1:200; Cat# GB23303; Servicebio, Wuhan, China). Finally,the brain sections were stained with the chromogenic agent 3,3-diaminobenzidine.
The number of viable surviving cells was measured as the number of cells per millimeter (1-mm) length in brain slices via light microscopy (BX40; Olympus, Tokyo, Japan)by two investigators who were blinded to the experimental conditions. Digitized images were obtained using a light microscope and quantified using ImageJ analysis software.
Electron microscope observation
Twenty-four hours after HIBD, neonatal rats in each group were anesthetized with anhydrous 2-butanol, and the cerebral cortex was removed and cut into 1 mm3pieces. The pieces were quickly placed into a pre-cooled 2.5% glutaraldehyde solution and stored overnight in a refrigerator at 4°C. The brain tissue was fixed with a mixture of 4% paraformaldehyde solution and 0.5% glutaraldehyde, processed by embedding,sectioning, and staining as previously described (Zhang et al., 2019), and then detected using transmission electron microscopy (Olympus).
Statistical analysis
Statistical analysis was performed with Prism 5 (GraphPad Software, San Diego, CA, USA). All data are presented as the mean ± standard error of the mean (SEM) and evaluated with one-way analysis of variance with Bonferroni’s multiplecomparisonspost hoctest. A value ofP< 0.05 indicates statistical significance.
Results
TIGAR exerts a protective effect in neonatal HIBD models in vivo and in vitro
First, to explore whether TIGAR expression was induced in the neonatal rat cortex by HIBDin vivo, we detected the TIGAR level after HIBD. As shown in Figure 1A, TIGAR expression was increased in the neonatal rat cortex after HIBD, reaching a peak level at 24 hours (P< 0.05,P< 0.01, orP< 0.001). Next, to assess the role of TIGAR in neonatal HIBD rats, we performed LV-sh_TIGAR-mediated knockdown of TIGAR in neonatal rat brains. The results showed that TIGAR knockdown by LV-sh_TIGAR decreased TIGAR expression (P< 0.01) and markedly exacerbated the brain lesion fraction 24 hours after HIBD compared with the model group (P<0.001; Figure 1B and C). Meanwhile, TIGAR expression was increased in OGD/R-induced HAPI microglial cells (P< 0.05 orP< 0.001; Figure 1D). Moreover, TIGAR knockdown by LV-sh_TIGAR not only significantly decreased TIGAR expression in HAPI microglial cells (P< 0.001; Figure 1E) but also aggravated cell viability and lactate dehydrogenase release (P< 0.01 orP< 0.001; Figure 1F and G). These results suggested that TIGAR exhibited a protective role in neonatal HIBD.
Pyroptosis is activated and increased by TIGAR knockdown in the cerebral cortex and HAPI microglial cells after neonatal HIBD modeling
To explore the effect of TIGAR on HIBD-induced activation of pyroptosisin vivo, we first examined the expression of pyroptosis-related proteins in the neonatal cortex after HIBD.The levels of caspase-1, GSDMD-N, and IL-1β were markedly increased after HIBD and peaked 24 hours after HIBD (P<0.05,P< 0.01, orP< 0.001; Figure 2A). Meanwhile, GSDMD knockdown in neonatal brains by injecting LV-sh_GSDMD significantly reduced infarction lesions 24 hours after HIBD compared with the model group (P< 0.001; Figure 2B).Moreover, thein vitrolevels of caspase-1, GSDMD-N, and IL-1β in HAPI microglial cells were markedly increased 24 hours after OGD/R (P< 0.05,P< 0.01, orP< 0.001; Figure 2C),and the levels of these proteins were further elevated by knockdown of TIGAR by LV-sh_TIGAR 24 hours after OGD/R (P< 0.01 orP< 0.001; Figure 2D).
TIGAR knockdown increases microglial pyroptosis through increasing ROS in vitro
Next, we explored the underlying mechanism of TIGARmediated pyroptosis in HAPI microglial cells after OGD/Rin vitro. The results showed that TIGAR knockdown with LV-sh_TIGAR significantly accelerated OGD/R-induced cell death and lactate dehydrogenase release, and the effects were alleviated by NADPH treatment (P< 0.01 orP< 0.001; Figure 3A and B).OGD/R markedly decreased the NADPH level and the GSH/GSSG ratio (Figure 3C and D) and increased ROS levels (Figure 3E). This effect was further aggravated by TIGAR knockdown with LV-sh_TIGAR (P< 0.05 orP< 0.001) but was reduced by NADPH treatment (P< 0.05,P< 0.01, orP< 0.001). Moreover,TIGAR knockdown by LV-sh_TIGAR observably elevated the increase in GSDMD-N and IL-1β levels induced by OGD/R(P< 0.01 orP< 0.001), while this was prevented by NADPH treatment (P< 0.001; Figure 3F).
TIGAR knockdown-aggravated neonatal brain injury after HIBD is suppressed by NADPH
We further investigated the influence of the PPP product NADPH on the protective effects of TIGAR in neonatal rat HIBDin vivo. The results showed that the lesion infarcts induced by TIGAR knockdown by LV-sh_TIGAR 24 hours after HIBD were markedly alleviated by NADPH administration (P< 0.05,P< 0.01, orP< 0.001; Figure 4A). Meanwhile, TIGAR knockdown by LV-sh_TIGAR obviously reduced the number of surviving cells in the neonatal rat cortex compared with the model group (P< 0.01), and this effect was blocked by NADPH administration 24 hours after HIBD (P< 0.05 orP<0.01; Figure 4B). Moreover, electron microscopy showed that TIGAR knockdown by LV-sh_TIGAR aggravated the formation of membrane pores and cell membrane rupture compared with the model group; however, this effect was reduced by NADPH administration (Figure 4C). Furthermore,immunohistochemical staining showed that GSDMD-N immunopositivity was upregulated in the neonatal rat cortex 24 hours after HIBD, and this effect was further elevated by TIGAR knockdown by LV-sh_TIGAR (P< 0.01) but prevented by NADPH administration (P< 0.05; Figure 4D).
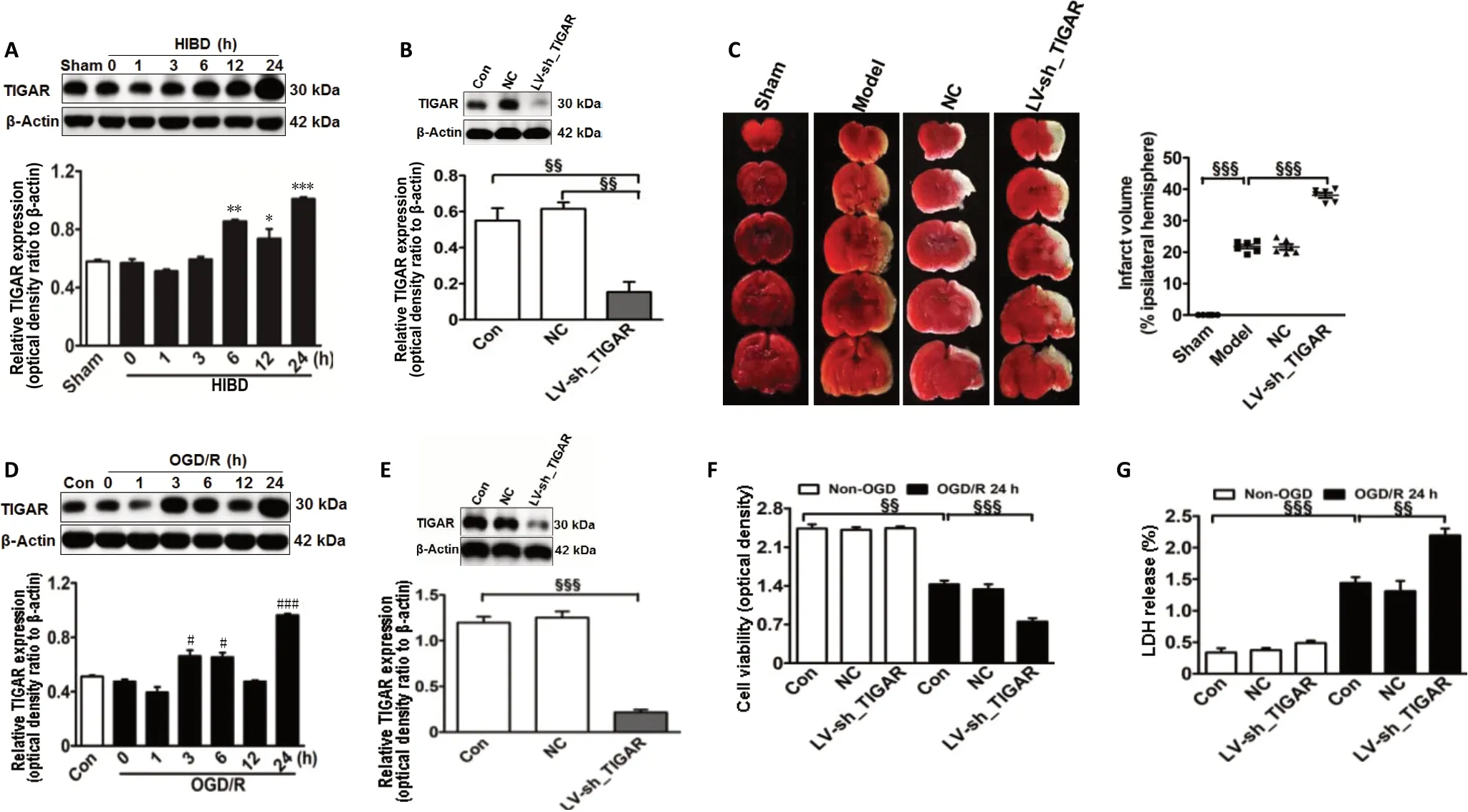
Figure 1 |TIGAR exerts a protective effect on neonatal HIBD models in vivo and in vitro.
Discussion
In the present study, we demonstrated the critical role of TIGAR in neonatal HIBD. Our studies found that TIGAR was increased in the HIBD-induced neonatal rat cortexin vivoand in OGD/R-induced HAPI microglial cellsin vitro. TIGAR knockdown in the neonatal rat brain significantly increased the infarct lesion after HIBD, and HAPI microglial cells exposed to OGD/R exhibited significantly increased pyroptosis. The detrimental effects of TIGAR knockdown in neonatal rat HIBD were blocked by the NADPH administration. These studies suggest that TIGAR prevents microglial pyroptosis and protects against neonatal HIBD.
Microglia play a crucial role in mediating the inflammatory response after HIBD (Kumari et al., 2010; Shen et al., 2010).Pyroptosis, a novel type of programed cell death associated with inflammation, exerts an important role in the process of ischemic brain damage (She et al., 2019). We previously demonstrated that TIGAR suppressed the astrocyte-mediated inflammatory response by inhibiting activation of nuclear factor-κB (Chen et al., 2018), and inhibition of nuclear factorκB transcription suppressed proinflammatory responses by decreasing activation of the NLRP-3 inflammasome (García et al., 2015). Recently, we found that inhibition of the NLRP-3/caspase-1/GSDMD axis reduced microglial pyroptosis and then alleviated neonatal HIBD (Lv et al., 2020). Therefore, we assumed that TIGAR might have a crucial role in microglial pyroptosis. In the present study, we found that OGD/R could induce the activation of microglial pyroptosis, as evidenced by the increased expression of caspase-1, GSDMD-N, and IL-1β in HAPI microglial cells, and these effects were reduced by TIGAR knockdown. These results imply that TIGAR prevents microglial pyroptosis in response to HIBDin vitro.
To elucidate whether TIGAR could play a protective role in HIBD, the present study explored the effect of TIGAR on neonatal rat HIBDin vivo. The results showed that TIGAR knockdown markedly increased the infarct lesion and pyroptosis after HIBD, which suggests that TIGAR protects against neonatal rat HIBD.
ROS are powerful initiators of neuroinflammation (Bok et al.,2017). Excessive intracellular ROS can trigger inflammatory responses and lead to cell death (Iizumi et al., 2016). TIGAR exhibits an anti-apoptotic effect by reducing the ROS levels through increasing the flux of the PPP (Bensaad et al., 2006).Previously, we found that TIGAR protected against ischemic brain injury by elevating the PPP flux and decreasing ROS levels (Li et al., 2014). Recently, we further demonstrated that NADPH (a metabolic product of the PPP) reduced ischemia/reperfusion-induced brain injury, dependent on its antioxidant and anti-inflammatory effects (Li et al., 2016).In the present study, TIGAR knockdown decreased the NADPH levels and GSH/GSSG ratio and increased the ROS levels in OGD/R-induced HAPI microglial cells. These effects were ameliorated by the administration of NADPH in vitro.Furthermore, NADPH administration also markedly blocked the detrimental effects of TIGAR knockdown on neonatal rat HIBD in vivo, which suggests that TIGAR reduces HIBD by increasing NADPH.
In conclusion, this study demonstrated that TIGAR suppresses microglial pyroptosis and protects against neonatal HIBD. The protective effect of TIGAR may be associated with an increase in NADPH. Thus, this study may provide a novel therapeutic target to treat neonatal HIBD.
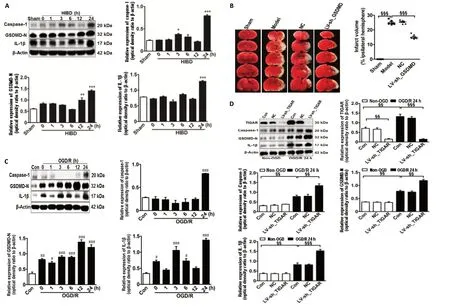
Figure 2 |TIGAR knockdown increases the activation of pyroptosis in the neonatal cortex and HAPI microglial cells of HIBD models in vivo and in vitro.
Author contributions:Study design: ML; animal and cell models establishment and drug treatment: LLT, XLJ; lesion infarct detection and hematoxylin and eosin staining: LXX, XD; western blot analysis: CXF,ZBZ; data analysis: GL, BS; manuscript writing: ML, ZHQ, XF; manuscript revision: ZHQ, XF. All authors approved the final version of this paper.
Conflicts of interest:The authors declare that they have no conflict of interests.
Financial support:This work was supported by the National Natural Science Foundation of China, Nos. 81872845 (to ML), 81771625 (to XF); the Natural Science Foundation of Jiangsu Province of China, No.BK20180207 (to ML); Jiangsu Provincial Medical Youth Talent of China,No. QNRC2016762(to ML); the Pediatric Clinical Center of Suzhou City of China, No. Szzx201504 (to XF); Postgraduate Research & Practice Innovation Program of Jiangsu Province of China, No. KYCX19_1998 (to LLT); Jiangsu Government Scholarship for Overseas Studies of China, No.JS-2017-127 (to ML); and the Fifth Batch of Gusu Health Talent Plan of China (to ML). The funders had no roles in the study design, conduction of experiment, data collection and analysis, decision to publish, or preparation of the manuscript.
Institutional review board statement:The study was approved by the Animal Ethics Committee of Soochow University (approval No.2017LW003) in 2017.
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement:Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix,tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Additional file:
Additional file 1:Original data of the experiment.
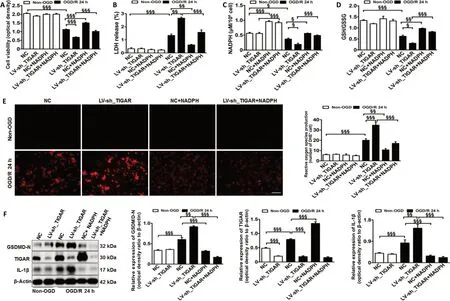
Figure 3 |TIGAR knockdown promotes microglial pyroptosis by increasing reactive oxygen species in vitro.

Figure 4 |Administration of NADPH reduces brain injury induced by TIGAR knockdown after neonatal HIBD.
- 中國神經(jīng)再生研究(英文版)的其它文章
- Entacapone promotes hippocampal neurogenesis in mice
- Electroacupuncture improves learning and memory functions in a rat cerebral ischemia/reperfusion injury model through PI3K/Akt signaling pathway activation
- Normobaric oxygen therapy attenuates hyperglycolysis in ischemic stroke
- MicroRNA-670 aggravates cerebral ischemia/reperfusion injury via the Yap pathway
- Corticospinal excitability during motor imagery is diminished by continuous repetition-induced fatigue
- Apelin-13 inhibits apoptosis and excessive autophagy in cerebral ischemia/reperfusion injury