
Motor neuron replacement therapy for amyotrophic lateral sclerosis
2022-01-12 04:31BochaoLiuMoLiLingyanZhangZhiguoChenPaulLu
中國神經(jīng)再生研究(英文版) 2022年8期
Bochao Liu, Mo Li, Lingyan Zhang, Zhiguo Chen, Paul Lu
Abstract Amyotrophic lateral sclerosis is a motor neuron degenerative disease that is also known as Lou Gehrig’s disease in the United States, Charcot’s disease in France, and motor neuron disease in the UK. The loss of motor neurons causes muscle wasting, paralysis,and eventually death, which is commonly related to respiratory failure, within 3–5 years after onset of the disease. Although there are a limited number of drugs approved for amyotrophic lateral sclerosis, they have had little success at treating the associated symptoms, and they cannot reverse the course of motor neuron degeneration. Thus, there is still a lack of effective treatment for this debilitating neurodegenerative disorder. Stem cell therapy for amyotrophic lateral sclerosis is a very attractive strategy for both basic and clinical researchers, particularly as transplanted stem cells and stem cell-derived neural progenitor/precursor cells can protect endogenous motor neurons and directly replace the lost or dying motor neurons. Stem cell therapies may also be able to re-establish the motor control of voluntary muscles. Here, we review the recent progress in the use of neural stem cells and neural progenitor cells for the treatment of amyotrophic lateral sclerosis.We focus on MN progenitor cells derived from fetal central nervous system tissue,embryonic stem cells, and induced pluripotent stem cells. In our recent studies, we found that transplanted human induced pluripotent stem cell-derived motor neuron progenitors survive well, differentiate into motor neurons, and extend axons into the host white matter, not only in the rostrocaudal direction, but also along motor axon tracts towards the ventral roots in the immunodeficient rat spinal cord. Furthermore, the significant motor axonal extension after neural progenitor cell transplantation in amyotrophic lateral sclerosis models demonstrates that motor neuron replacement therapy could be a promising therapeutic strategy for amyotrophic lateral sclerosis, particularly as a variety of stem cell derivatives, including induced pluripotent stem cells, are being considered for clinical trials for various diseases.
Key Words: amyotrophic lateral sclerosis; motor neuron replacement; neural progenitor cells; neural stem cells; stem cells
Introduction
Amyotrophic lateral sclerosis (ALS), one of the most common progressive neurodegenerative diseases, was first identified by Sir Charles Bell in 1830, and was fully characterized by Jean-Martin Charcot in 1874. The disease is also known as Lou Gehrig’s disease in the USA, Charcot’s disease in France, and motor neuron (MN)disease in the UK. The average incidence rate of ALS worldwide is about 1/50,000 per year, which equates to more than 100,000 new diagnoses per year (Norris et al., 2020). The loss of MNs can occur within both the lower spinal cord and upper cortical circuits. Because these neurons transmit signals between the brain and the voluntary muscles, the loss of MNs can lead to deterioration of motor control.
Symptoms include progressive muscle wasting, paralysis, eating and breathing difficulties, and muscle atrophy, all of which eventually lead to death related to respiratory failure within 3–5 years of disease onset (Atassi et al., 2016; Chen et al., 2016). A small fraction (~5–10%)of ALS cases are familial, and are associated with pathogenic gene mutations in C9ORF72, Cu/Zn superoxide dismutase 1 (SOD1), TAR DNA-binding protein, FUS and ubiquilin 2. However, the vast majority(over 90%) of ALS cases are considered sporadic, and are the result of genetic load, aging and environmental exposure in susceptible individuals (Hardiman et al., 2017; van Es et al., 2017).
At present, no treatment is available to reverse the pathological progression of ALS. Two drugs with very modest effect on survival have been approved by the US Food and Drug Administration (FDA):Rilutek (riluzole) and Radicava (edaravone). Rilutek was approved by the FDA in 1995 and has been listed in Canada, Australia, Europe and other countries. Rilutek is thought to interfere with the activity of glutamate, one of the chemical messengers that transmit signals between nerve cells. Excess glutamate can be toxic in the brain and spinal cord, as in ALS (Mignani et al., 2020; Calabrese et al., 2021).Radicava was approved by the FDA in 2017 and by Health Canada in 2018, based on findings from Study 19 (2011-NCT01492686), a double-blind phase 3 clinical trial in Japan that assessed the safety and efficacy of Radicava in people with ALS. In this clinical trial,participants on Radicava experienced a significantly slower decline(by 33%) in their ability to perform everyday activities, compared with those given placebo. Currently, the treatment is only available in the United States, Canada, Japan and South Korea, although the European drug administration is also considering its use. Radicava is thought to counteract the excessive oxidative stress in ALS (Jackson et al., 2019).
Stem cell therapy for ALS continues to be a hot topic in both basic and clinical research. No current drug or non-drug treatment for ALS has stimulated as much anticipation as stem cells. Neuroprotection is one of the major objectives of stem cell therapy in ALS clinical trials. Stem cells can provide immunomodulation, secrete growth factors, and produce supporting cells, such as astrocytes and oligodendrocytes, or interneurons, which may provide a supportive environment to protect damaged MNs against degeneration (Atassi et al., 2016; Berry et al., 2019). Neuronal replacement is another promising therapeutic strategy for ALS. Here, the transplanted stem cells or stem cell-derived neural progenitor cells can directly replace the dying or dead MNs in the host and re-establish the reciprocal connections to restore the motor control of voluntary muscles in ALS (Abati et al., 2019b; Forostyak et al., 2020). However, for ALS,there are still no approved stem cell therapies, although there are a few ongoing stem cell clinical trials (2021-NCT02478450, 2020-NCT03296501, 2018-NCT03482050). In this review, we will cover the progress and results of these trials using stem/progenitor cells derived from a variety of sources. We will also review our own work to provide a historical perspective on how and why stem cells are considered a potential treatment option for patients with this incurable neurodegenerative disease.
The challenges in this approach revolve around the delivery and source of stem cells. Obviously, the location of stem cell delivery will affect the therapeutic effect. Several preclinical studies have reported transplantation of glial cell line-derived neurotrophic factor(GDNF)-secreting neural progenitor cells into SOD1G93Arats. In one study, neural progenitor cells were injected into four unilateral sites in the lumbar L1/L2 spinal cord in rats at the pre-symptomatic stage(70 days of age). Surprisingly, despite the survival of some MNs 2–6 weeks after transplantation, neither MN–muscle contact nor improvements in ipsilateral hind limb function could be observed(Suzuki et al., 2007). In contrast, transplantation protects MNs and respiratory function when GDNF-secreting neural progenitor cells are injected into four unilateral sites at the cervical C3 and C6 spinal cord levels in the early to mid-symptomatic stage at 110–120 days of age (Nichols et al., 2013; Zalfa et al., 2019). In addition to spinal cord transplantation, human cortical-derived GDNF-secreting neural progenitor cells injected into 20 sites in the motor cortex of SOD1G93Arats at the pre-symptomatic stage at ~80 days of age prevents degeneration of spinal MNs, delays disease pathology, improves function, and increases life span (Thomsen et al., 2018). With the development of new stereotactic devices, it is feasible to transplant neural precursor cells into the brain parenchyma. Nonetheless,the best transplant routes for ALS treatment need to be carefully examined. In future cell therapy studies, the impact of the delivery route should be carefully assessed.
Over the past few decades, various cell sources such as neural stem cells (NSCs) isolated from embryonic central nervous system tissue (brain and spinal cord), mesenchymal stem cells (MSCs),hematopoietic stem cells and embryonic stem cells (ESCs) have been employed to treat ALS patients (Figure 1). Recently, induced pluripotent stem cells (iPSCs) derived from adult human tissues have become popular (Yu et al., 2007). iPSC-derived neural stem/progenitor cells could greatly reduce, if not completely avoid, the immune rejection and ethical concerns associated with the use of fetuses or embryonic tissues (Figure 1). Furthermore, human iPSCderived NSCs can survive and potentially differentiate into neurons and glia after transplantation into the spinal cord in both rat and mouse models (Sareen et al., 2014). Therefore, they provide the possibility of autologous transplantation in clinical trials.
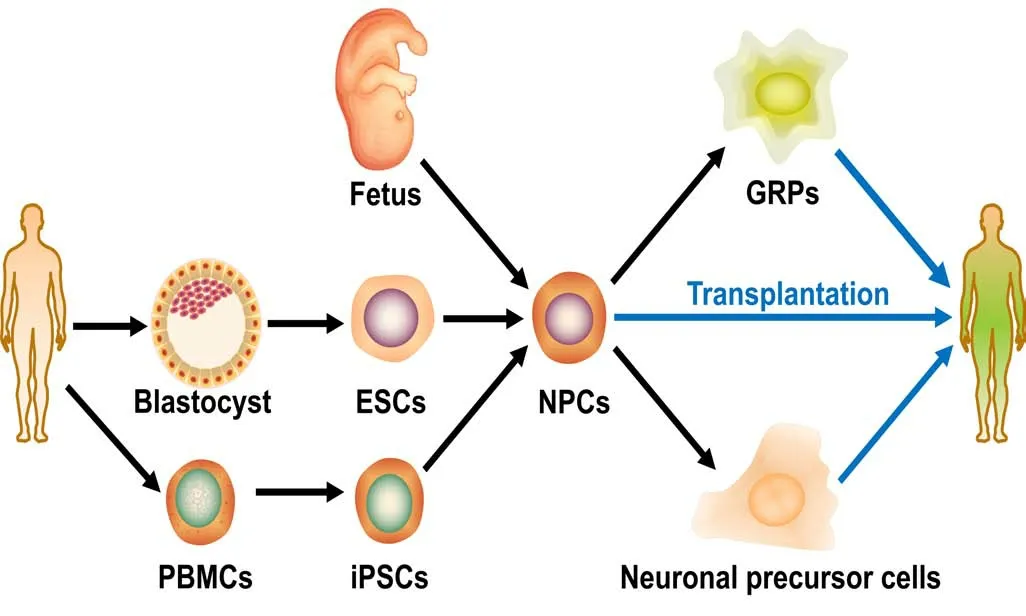
Figure 1 |Cellular sources for transplantation to treat amyotrophic lateral sclerosis.Three major steps are included in the process of cell transplantation—cell extraction, differentiation and transplantation. Neural progenitor cells(NPCs) can be either isolated from fetuses or from other stem cells such as embryonic stem cells (ESCs, derived from the inner cell mass of blastocysts)or induced pluripotent stem cells (iPSCs, reprogrammed from peripheral blood mononuclear cells (PBMCs)). The NPCs can be directly used for transplantation or following one more step that specifies the cells to lineagespecific precursor cells, such as glial-restricted precursors (GRPs) and neuronal precursor cells.
Fetal Central Nervous System-Derived Neural Stem/Progenitor Cells
Fetal human CNS tissue contains a population of neural stem/progenitor cells primed to differentiate into neurons, astrocytes and oligodendrocytes (Lyman et al., 1991; Mazzini et al., 2015;Ferrari et al., 2018). In 1977, the first evidence was presented that grafts of fetal brain tissue into the adult CNS could counteract an experimentally-induced neurological deficit (Nygren et al., 1977).Then, in 1983, the first successful transplantation of fetal spinal cord into the adult spinal cord was reported (Patel and Bernstein, 1983).After the success of fetal CNS tissue-based transplantation, NSCs were isolated from fetal CNS tissues and culturedin vitro. By using growth factors such as basic fibroblast growth factor (bFGF) and epidermal growth factor (EGF), fetal CNS-derived neural stem/progenitor cells could be maintained and passaged long-term without losing stemness (Shihabuddin et al., 1999). In animal studies,these cells were shown to differentiate mostly into interneurons and less frequently into astrocytes that release numerous growth factors (Suzuki et al., 2007; Nichols et al., 2013). It was also reported that fetal-derived stem/progenitor cells originally isolated from neurogenic and non-neurogenic zones could be delivered into the spinal cord to protect MNs in animal models (Baloh et al., 2018).
The generation of cholinergic MNs in MN replacement therapy is considered of fundamental importance in ALS treatment. In an early study published in 2002, Wu et al. reported a newin vitropriming procedure that involves treatment of NSCs (K048) derived from the cortex of an 8-week legally-aborted human fetus with numerous chemical compounds and growth factors, including bFGF, EGF,leukemia inhibitory factor, mouse sonic hedgehog amino-terminal peptide, all-trans retinoic acid, NGF, BDNF, NT-3, NT-4, natural mouse laminin and heparin (Wu et al., 2002). With this manipulation, almost all fetal brain-derived NSCs differentiate into a nearly pure population of neurons after grafting into the adult rat CNS. Furthermore, the transplanted cells differentiate into cholinergic MNs in a regionspecific manner, particularly in the spinal cord ventral horn, an area where MNs are lost in ALS patients. These studies have brought neuronal replacement therapy one step closer to being a potential candidate to treat ALS in upcoming clinical trials.
In another study, human glial-restricted progenitor cells (hGRPs)were tested in both transgenic rat and mouse ALS models carrying the hSOD1G93Amutation for MN protection (Howland et al., 2002;Lepore et al., 2011). In this case, specific growth factors such as bFGF and platelet-derived growth factor-AA were used in the priming procedure. With this approach, fetal brain-derived cells lineagespecifically differentiate into GRPs that can further differentiate into astrocytes and oligodendrocytes. After transplantation of hGRPs into the cervical spinal cord of hSOD1G93Amice, the cells survived and migrated in both gray and white matter, and differentiated into astrocytes. However, the transplantation neither protected MNs nor improved motor function (Lepore et al., 2011). In contrast, in a paper previously published by the same group, several therapeutic effects, including MN protection, delay in respiratory failure and slowed disease progression, were reported for rat-derived GRP-based therapy in the hSOD1G93Arat model (Lepore et al., 2008). Notably,these two GRP-based strategies share common features, including comparable cell types, delivery routes and intervention times (6 or 4 bilateral injection sites at C4–C6 or C4–C5 ventral horn in the hSOD1G93Arat or the hSOD1G93Amouse at 90 or 50–60 days of age).This suggests that the immune response may play a critical role in the effectiveness of these therapeutic approaches, particularly as one is a xenograft and the other is an allograft. Nevertheless, the therapeutic potential of GRPs is significant, as is evidenced by the key role of glial cells in disease and neuronal survival in SOD1 ALS animal models.
Several trials using expanded human fetal-derived stem/progenitor cells to treat ALS have been completed in Europe (Mazzini et al.,2015; Zalfa et al., 2019) and the United States (Glass et al., 2012,2016; Mazzini et al., 2015). Importantly, none of these trials were intended to replace lost MNs, which would be impractical because the new MNs would need to connect to distant target muscles.Instead, the investigators aimed to replace the interneurons and astrocytes surrounding the dying MNs, release potent growth factors into degenerated or degenerating areas, and/or provide immunomodulatory effects to reduce toxic inflammation or some other detrimental process. In addition to modulating the microenvironment, another rationale of neuronal cell transplantation is that once new synaptic connections are established between engrafted neurons and host degenerating MNs, the enhanced electrophysiological activity afforded by the newly-formed synapses may slow down the pathological degeneration of MNs in ALS. The largest among these trials was a phase 1 and 2 trial of intraspinal transplantation of cells derived from human fetal spinal cordtissue, with 15 participants in three centers, and was supported by Neuralstem Inc. and the National Institute of Neurological Disorders and Stroke in the USA (Glass et al., 2016).
In one study, NSCs/progenitor cells were extracted from the brain of an 8-week-old fetus, amplified in culture, and frozen in varying doses.MNs were successfully induced from these CNS-derived progenitor cells (Guo et al., 2010). Another source is the fetal cortex, which generates protective astrocytes when transplanted into the spinal cord of rodents. These cells have been genetically engineered to express GDNF, which has a protective effect on MNs in animal models of ALS (Suzuki et al., 2007; Nichols et al., 2013). This combined gene therapy strategy is currently being evaluated in a single-center phase 1 and 2a dose-escalation clinical trial in California (2017-NCT02943850).In this trial, Baloh et al. (2018) transplanted cells into the lumbar spine in 18 patients (as of October, 2019). The primary outcome measures are safety and tolerability.
A phase 3 study (2017-NCT03280056) was recently completed on October 30, 2020. In this trial, 261 participants were recruited at six U.S. sites to evaluate the efficacy and safety of BrainStorm Cell Therapeutics’ candidate NurOwn, which is an autologous neurotrophic factor-secreting mesenchymal stromal cell. In February,2021, the FDA concluded in a preliminary review that the available safety and efficacy data from the phase 3 clinical trial of NurOwn were insufficient to support approval of the treatment. Nevertheless,NurOwn is still considered one of the most promising therapeutic candidates for clinical application for ALS.
Despite the substantial progress over the past few years, cell therapies based on fetal CNS-derived NSCs/progenitor cells must still overcome a number of limitations before clinical application. These include relatively high immunogenicity, low engraftment success and low survival rate after transplantation. In addition, it remains a challenge to generate phenotype-specific neuronal progenitor cells,such as MNs and MN-associated interneurons, for MN replacement therapy. Moreover, ethical concerns and the limited supply of human fetal tissue are additional hurdles to clinical translation.
Embryonic Stem Cell-Derived Neural Stem/Progenitor Cells
ESCs show unlimited capacity for self-renewal and can be expanded indefinitely in culture. Notably, ESCs have the potential to differentiate into any cell type of the three germ layers, including NSCs with neuronal or glial fates. Therefore, they have great therapeutic potential for tissue replacement after injury or disease,such as in regenerative medicine or replacement of MNs in ALS.However, it remains controversial whether MNs derived from ESCs can exert beneficial effects in animal models of ALS (Deshpande et al., 2006; Lopez-Gonzalez et al., 2009). Because of the ethical concerns as well as difficulties in generating high-purity lineage/fatespecific cell lines with tolerable risk of tumorigenesis, the clinical application of ESCs is still limited.
In 2004, Harper et al. generated spinal MNs from mouse ESCs to evaluate the developmental potential of these cellsin vitro, and they further examined their capacity to replace MNs in the adult mammalian spinal cord. In this study, 5–7-week-old male Lewis rats were given intracranial injection of rat-adapted neuroadapted Sindbis virus (NSV) to damage the resident MNs. These rats then served as hosts for transplant of MN-fated mouse ESCs into the spinal cord.One month later, over 3000 mouse ESC-derived MNs (~25% of input)survived (Harper et al., 2004). However, transplant-derived neurons extend very few axons into the host white matter. An inhibitory effect of white matter myelin on ESC-derived neuronal axonal growth has been reported. This result, however, is not consistent with our own observations. In our recent studies, we reported enhanced axonal growth in adult host white matter (Lu et al., 2012, 2014, 2017;Poplawski et al., 2018). Nevertheless, the inhibitory effect of myelin can be overcome by treatment with dibutyryl cAMP (dbcAMP) or Y27632 (a Rho kinase inhibitor) bothin vitroandin vivo. Importantly,in the dbcAMP-treated group, but not in the non-dbcAMP-treated group, ~80 ESC-derived motor axons extended into the ventral roots of the spinal cord (Harper et al., 2004). These results suggest that ESC-derived neural stem/progenitor cells have the capacity to replace lost or dying MNs in ALS and other MN degeneration-associated diseases.
To investigate whether transplanted MNs exert a beneficial effect in MN degenerative diseases by replacing host MNs or preventing their degeneration, MNs differentiated from mouse ESCs were transplanted into the lumbar spinal cord of wild-type (WT) and hSOD1G93Arats at 10 weeks, i.e., during the pre-symptomatic stage.MNs were identified by expression of a green fluorescent protein(GFP) marker under control of the promoter forhb9, a MN-specific gene (Lopez-Gonzalez et al., 2009). These motor neuronal-like grafted cells can survive for at least 1 week in hSOD1G93Aanimals.However, neither grafted GFP+neurons nor the endogenous choline acetyl transferase (ChAT)+/Hoechst–/GFP–MNs survived in either sham or grafted SOD1G93Arats in the long-term. In contrast, in WT rat spinal cords of the same age, grafted GFP+MNs were detected. The loss of both host and transplanted MNs is correlated with a sudden decrease in motor performance from week 16 onwards. These results indicate that the environment in transgenic hSOD1G93Aanimals is antagonistic to both endogenous and grafted MNs, raising the concern of whether the direct replacement of lost MNs can reverse the pathological progression of ALS (Lopez-Gonzalez et al., 2009).
Application of Induced Pluripotent Stem Cells Derivatives in the Treatment of Amyotrophic Lateral Sclerosis
Cell therapy for ALS works mainly through cell replacement or supportive effects to delay the progression of ALS. As described above,numerous animal and clinical studies of ALS have been carried out.Cell intervention therapy seems to have positive effects for ALS, but obtaining a stable and sufficient cell source is still a prerequisite for cell therapy in clinical trials. NPCs derived from embryonic neural progenitors or differentiated from ESCs are the only cell type that cannot avoid the limitation of cell numbers or avoid ethical concerns.Obtaining enough cells for transplantation is still a key challenge in the field. The generation of iPSCs reprogrammed from somatic cells(Takahashi et al., 2007; Yu et al., 2007; Park et al., 2008) has afforded an opportunity for the development of autologous cell replacement therapy (Robinton and Daley, 2012; Yamanaka, 2012; Okano et al.,2013; Glicksman, 2018). With the development of transdifferentiation manipulation, the starter cell types and reprogramming methods have progressed (Loh et al., 2009; Malik and Rao, 2013; Isogai et al., 2018),the differentiation efficiency has been improved, and the generation of iPSCs has been made simpler. iPSCs are essentially infinitely proliferative and can be stored and differentiated into any type of cell. This feature solves the cell source problem and provides an unprecedented opportunity for autologous transplantation. However,obtaining autologous iPSCs from patients takes a long time and is costly. To overcome these difficulties, several groups are currently trying to bank clinical-grade iPSC lines from human leukocyte antigen homologous donors (Taylor et al., 2005; Azuma and Yamanaka, 2016),and clinical trials are also being carried out (Morizane, 2019).
Although iPSCs are a promising cell type for cell replacement therapy, they carry an enormous risk of tumor formation. The risk of tumorigenesis is associated with three key factors. The first is that cell differentiation may not be complete. After the undifferentiated/immature cells are transplanted, they may undergo tumorigenesis.Second, because the reprogramming factors may remain active in iPSCs, they may promote the occurrence of tumor. Third,tumorigenesis may be caused by genetic mutations that occur during the reprogramming or culture processes (Yamanaka, 2020).Accordingly, numerous studies have been carried out to improve iPSC reprogramming technology, and the emergence of new nonintegrated methods, animal component-free culture and innovative virus-free strategies have greatly reduced the risk of tumorigenesis and enhanced the therapeutic applicability of iPSCs (Stadtfeld et al., 2008; Fusaki et al., 2009; Ban et al., 2011; Malik and Rao,2013; Hamada et al., 2020). Mandai et al. (2017) conducted the first clinical trial using iPSC derivatives. A patient with age-related macular degeneration received autologous iPSC-differentiated retinal pigment epithelial cells (iPSCs that were derived from the cutaneous fibroblasts of the patient) without administering an immunosuppressive regimen. After transplantation, neither signs of immune rejection nor abnormal genomic changes were observed,indicating that the cell graft did not form any tumors that could be detected. These results emphasize the great potential of iPSC technology as a new tool for cell therapy, and it may become a major strategy in cell therapy.
Degeneration and apoptosis of MNs are the main pathological hallmarks of ALS. Therefore, it is rational to contemplate treatment of ALS patients with MNs derived from ESCs or iPSCs. Unfortunately,studies have shown that this approach may be difficult to implement successfully (Lopez-Gonzalez et al., 2009; Boulis et al., 2011; Abati et al., 2019b). For example, researchers have transplanted mouse ESC-derived MNs into a mouse model of ALS, and found that while it improved the symptoms of ALS, the transplanted MNs failed to integrate into the host neural circuits under the antagonistic microenvironment (Lopez-Gonzalez et al., 2009). Further analysis showed that the transplanted MNs failed to replace the apoptotic neurons, and also failed to form synaptic connections with the host endogenous neurons and distal muscles. The grafted cells must be able to project neurites long distances and overcome the nonpermissive microenvironment. This may account for why ESC and iPSC-derived MN replacement therapy for ALS has not yet been successful. Thus, current stem cell-based therapy is still mainly focused on neuroprotection (Chen et al., 2016; Baloh et al., 2018).Transplanted NSCs secrete neurotrophic factors and preserve perineuronal nets to provide neuroprotection (Forostyak et al.,2020). They can also differentiate into glial cells, interneurons and MNs and play a supporting role to slow down the progression of the disease (Lunn et al., 2014; Goutman et al., 2019). In studies in which iPSC-derived NPCs were transplanted into the lumbar spinal cord of an ALS mouse model, the cells differentiated into neurons and astrocytes and significantly increased lifespan (Popescu et al., 2013;Kondo et al., 2014). However, this technology is still in the early stage of development, and iPSC derivatives have not yet been used in clinical trials for ALS.
Our Own Work on Amyotrophic Lateral Sclerosis Therapy
Du et al. (2015) reported the differentiation of MN progenitor (MNP)cells from human pluripotent stem cells. These progenitor cells can be passaged at least five timesin vitro. After the application of a NOTCH pathway inhibitor, more than 90% of the cells differentiated into MNs. This differentiation method can give rise to human MNs in a highly efficient manner, providing an opportunity for further research on ALS.MNs or neural progenitor cells derived from human iPSCs (iPSC-NPs),especially those derived from ALS patients, are mainly prepared for ALS disease modeling (Chen et al., 2014; Ilary Allodi, 2016). Few studies have attempted to transplant these MNs or their progenitor cells for replacement therapy (Zhu and Lu, 2020). Recently, Forostyak et al. (2020) reported the application of iPSC-NPs in SOD1 rats.They found that rat MNs were significantly preserved, disease progression was slowed down, and the survival of all treated animals was prolonged. Although iPSC-NPs could interact with rat MNs, they mostly remained in an undifferentiated stage. The results show that this cell type mainly plays a neuroprotective and regulatory role by regulating local gene expression or by remodeling the extracellular matrix in the CNS (Forostyak et al., 2020). Most ALS patients die of asphyxia due to end-stage respiratory failure caused by the lack of MN innervation of respiratory muscles. Given the shorter distance between the cervical spinal cord and the respiratory muscles, it may be relatively easier to re-establish the MN axonal projections to respiratory muscles by transplantation at the cervical spinal cord versus at other regions, such as the lumbar spinal cord. Further studies are warranted to address these issues.
Recently, iXCells Biotechnologies developed a robust method to produce highly pure, functionally-validated, and ready-to-use MNs from hiPSCs using NSC induction medium, MN progenitor differentiation medium, MN differentiation, and maintenance medium. These human iPSC-derived MNs or their progenitors can be used not only for neurological disease modeling and toxicology studies, but also have potential for MN replacement therapy for ALS. These MNs have greater than 85% purity as measured by immunostaining for the motor neuronal markers ISL1, HB9 and ChAT (Figure 2A). In addition, iXCellsTMhiPSC-derived MNs are functionally verified by neuromuscular junction assay (Figure 2B) and electrophysiological assays.
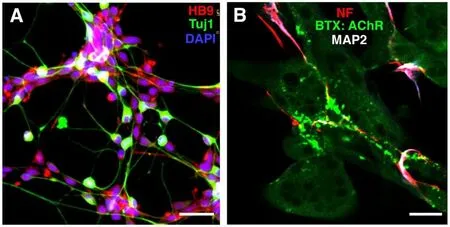
Figure 2 |Human induced pluripotent stem cells-derived iXCellsTM motor neurons.
In a joint effort, iXCells Biotechnologies is collaborating with the Lu lab and the Chen lab to test these cells in ALS animal models.We have obtained MNs derived from hiPSCs as well as SODG93A transgenic and WT monkeys, and transplanted hiPSCs-MN progenitor cells into the spinal cord ventral horn in immunodeficient rats. Nine rats received 0.25 μL of cells at a concentration of 100,000 cells/μL per site for a total of eight sites spanning 0.5 mm apart in both the right and left hemispheres at the C5 spinal cord using a pulled glass needle connected to a PicoSpritzer II (Lu et al., 2012). Animals survived 3 or 6 months post transplantation. Transplanted iXCellsTMhiPSC-derived MN progenitors labeled with GFP survived well in the host spinal cord, as verified by both GFP and human nuclei marker immunolabeling in a pilot study (Figure 3A). The engrafted cells could differentiate into mature ChAT+MNs (Figure 3B), indicating preservation of MN fate and continuous maturation of MNsin vivo.Furthermore, transplanted MNs extended into a large number of nerve fibers following the motor axon tracts along the white matter of the spinal cord (Figure 3C).
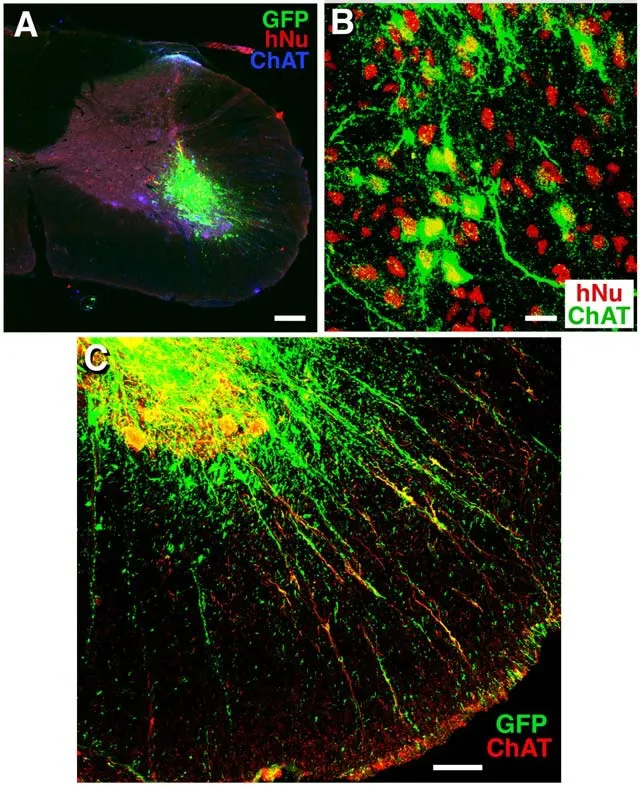
Figure 3 |Transplanted MNPs differentiate into motor neurons and extend their axons along motor axon tracts.(A, B) Transplanted human induced pluripotent stem cells (iPSCs)-derived motor neuron progenitors (MNPs)labeled for human nuclei(hNu) and GFP survived in the mid-cervical spinal cord ventral horn in immunodeficient rats(A) and differentiated into choline acetyl transferae (ChAT+) motor neurons 3 months post transplantation (B). (C)Transplant-derived neurons extended their axons along motor axonal tracts in the ventral spinal cord. Scale bars: 200 μm in A, 18 μm in B, and 68 μm in C.Unpublished data.
Notably, some of these nerve fibers were ChAT+, indicating that they are transplant-derived motor axons. In addition, some of these fibers reached the spinal cord ventral border and even extended into the ventral roots. In addition, transplanted hiPSC-derived MNs extended axons over a long distance towards the caudal direction in the host spinal cord (Figure 4). Furthermore, there was no tumor formation or any detectable expression of classical tumorigenic factors 6 months after transplantation, indicating good safety. We have also established ALS disease models of mice and monkeys. In the future,we will transplant hiPSC-MNs into these ALS animals to study the potential of MN replacement therapy for ALS.
Future Perspectives
The pathogenesis of ALS is not fully understood, and there are many pathogenic mechanisms that may affect the occurrence and development of the disease. Rodent studies have demonstrated that ALS may include a non-cell autonomous mechanism, with glial cells playing an important role in the onset and progression of ALS(Arbour et al., 2017; Rostalski et al., 2019; Izrael et al., 2020). Recent evidence suggests that both innate and adaptive immunity are also involved in the progression of ALS (Zhao et al., 2013; Rodrigues et al.,2014). Thus, stem cell therapy may not only need to replace dead or dying MNs, but also improve the ALS microenvironment to delay or even reverse disease progression. The hostile niche significantly exacerbates the degeneration of endogenous MNs and inevitably negatively impacts transplanted grafts as well. Therefore, a major challenge is enhancing the survival of transplanted MN precursors.
The survival rate of the transplanted cells can be improved by modifying the transplanted cells or improving thein vivomicroenvironment. For example, some anti-apoptotic genes (HIF1a and Bcl-2), antioxidants (SOD2, catalase and Nrf2) and trophic factors (VEGF, EPOR, GDNF and NT-3) can be upregulated by hypoxia,neurotrophic factors or small molecule drug preconditioning or gene engineering. Recently, researchers have encased graft cells in biomaterials to improve durability and performance in the host tissue after transplant (Abati et al., 2019a; Wang et al., 2020). To overcome the strong immunogenicity that potentially hinders the survival of transplanted cells, researchers are now developing stable induced NSCs (iNSCs) as well as MNP cells derived from iPSCs or through direct reprogramming of human blood mononuclear cells. These cells can be used to treat neurological diseases such as ALS (Rosati et al., 2018; Yuan et al., 2018). To overcome the adverse environment,combination therapy of MSCs and NSCs could also be a good choice for future clinical trials (Petrou et al., 2016; Berry et al., 2019).NSCs and MSCs are two promising cell types for potential clinical applications. The biological characteristics of MSCs and the results of clinical trials indicate that MSCs can be used as immunomodulators for ALS treatment, either through intravenous or intrathecal infusion(Bonafede and Mariotti, 2017; Tang, 2017; Gugliandolo et al., 2019).Furthermore, NSCs can be directly transplanted intraspinally to target degenerating MNs in the ventral horn. Implanted NSCs mainly differentiate into neurons and glia, such as astrocytes, which do not only ameliorate the toxic microenvironment surrounding MNs by releasing growth factors and immunomodulatory molecules, but may also replace degenerated MNs for the reconstruction of neural circuits (Abati et al., 2019a). For the application of MNP cells, the addition of niche-modulating cells, such as MSCs and astrocytes(Chandrasekaran et al., 2016; Izrael et al., 2018; Filipi et al., 2020),or co-transplantation of biomaterials carrying growth factors and neurotrophic factors (Moshayedi et al., 2016) might improve the efficacy of the treatment of ALS.
The second challenge is evaluating the engraftment of transplanted cells in clinical trials. The majority of published techniques to evaluate engraftment, such as immunofluorescent staining and fluorescent protein labeling, are difficult to implement in the clinical setting. In contrast to the lack of safe, non-invasive and time efficient technique(s) to monitor and assess the therapeutic efficacy of transplanted cells in ALS patients, molecular imaging techniques have been used for the tracking and evaluation of therapeutic outcomes in stem cell-based therapies (Klontzas et al., 2021) in cardiac diseases(Li and Hacker, 2017), Parkinson’s disease (Jang et al., 2020) and Alzheimer’s disease (Alipour et al., 2019; Klontzas et al., 2021).Thus, noninvasive imaging techniques such as magnetic resonance imaging, positron emission tomography and single photon emission computed tomography also have great potential to track the fate of transplanted cells in ALS patients.
The third challenge is establishing connectivity between transplanted MNs and remote downstream targets, such as muscles, glands and various organs, which requires extensive long-distance growth of motor axons. This is in contrast to development, where axons travel only for a short distance to reach their targets. Transplantation of MNs in the peripheral nerve close to their targets could be a shortcut for connectivity. Transplantation of interneurons in the peripheral nerve to relay the motor signals to their targets could be another strategy worthy of investigation.
The last challenge is selecting appropriate patients for enrollment.Ultimately, MN replacement therapy for ALS aims to protect endogenous MNs or even directly replace the lost or dying MNs.However, re-establishing these connections may require several months or even years. Thus, only patients with a medium or slow progression of disease should be included in the clinical trial. In addition, because of the strong placebo effect, diverse progression rates and subtypes, double-blind design is crucial for ALS trials. An extreme example is contributed by the antibiotic minocycline. Animal studies and open-label human trials have shown that minocycline is beneficial for ALS. However, a larger placebo-controlled trial suggests that it is not, and may even be harmful (Lou et al., 2010).As intraspinal transplantation is an invasive treatment, persuading patients to be included in a double-blind trial is always a challenge.As of the writing of this manuscript, the FDA granted investigational new drug trial approval for the use of ESC-derived dopaminergic neuronal precursor cells for the treatment of Parkinson’s disease (Piao et al., 2021; Takahashi, 2021) after the investigators showed rigorous safety and efficacy data in preclinical studies. This may herald the advent of a new era of cellular therapy for neurological diseases.ESCs, iPSCs and iNSCs can be differentiated into MNs and nichesupporting cells. After robust safety and efficacy testing, it is possible to see these cells being tested in a well-designed clinical trial in the near future.
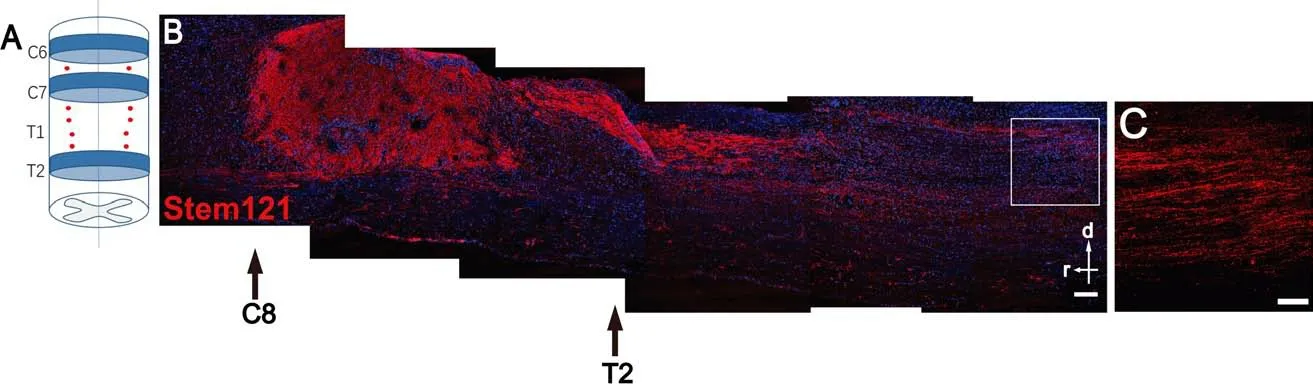
Figure 4 |Induced pluripotent stem cells-derived motor neurons (MNs) survive and extend axons in the host.(A) A diagram showing human induced pluripotent stem cells -derived MNs transplanted into 10 sites bilaterally between the C6–T2 vertebrae (red dots, transplant sites; T1 vertebra was removed by laminectomy). (B) MNs survived well in the spinal cord of adult immunodeficient rats 6 months after transplantation. Moreover, a large number of axons had projected to the caudal host spinal cord. (C) An enlarged view of the inset in B (red channel only) to show graft-derived axons in the caudal region. d: Dorsal; r: rostral;red: Stem121-labeled transplanted human cells; blue:4′,6-diamidino-2-phenylindole (DAPI); scale bars: 250 μm in B, and 100 μm in C. Unpublished data.
Author contributions:PL, ZC, and LZ designed the manuscript and finalized this review. BL and ML wrote the manuscript. All authors approved the final manuscript.
Conflicts of interest:The authors declare no conflicts of interest. No conflicts of interest exist between iXCells Biotechnologies USA, Inc. and publication of this manuscript.
Open access statement:This is an open access journal, and
articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
?Article author(s) (unless otherwise stated in the text of the article) 2022. All rights reserved. No commercial use is permitted unless otherwise expressly granted.
- 中國神經(jīng)再生研究(英文版)的其它文章
- Calorie restriction or dietary restriction: how far they can protect the brain against neurodegenerative diseases?
- Monodelphis domestica: a new source of mammalian primary neurons in vitro
- Vascular inflammation in the central nervous system
- Proper progression of neurogenesis relies on a defined pattern of SUMOmodified proteins
- Elucidating the pathological mechanisms of neurodegeneration in the lethal serpinopathy FENIB
- Glial cell line-derived neurotrophic factor in brain repair after focal ischemic stroke