
Molecular simulation studies on natural gas hydrates nucleation and growth: A review
2022-05-24 09:45:32ZhengciZhngNengyouWuChnglingLiuXiluoHoYongchoZhngKiGoBoPengChoZhengWeiTngGungjunGuo
China Geology 2022年2期
Zheng-ci Zhng, Neng-you Wu, , Chng-ling Liu, , Xi-luo Ho, , Yong-cho Zhng, , Ki Go ,Bo Peng , Cho Zheng , Wei Tng , Gung-jun Guo ,*
a Laboratory for Marine Mineral Resources, Pilot National Laboratory for Marine Science and Technology, Qingdao 266071, China
b The Key Laboratory of Gas Hydrate, Ministry of Natural Resources, Qingdao Institute of Marine Geology, Qingdao 266071, China
c Key Laboratory of Petroleum Resource Research, Institute of Geology and Geophysics, Chinese Academy of Sciences, Beijing 100029, China
d College of Earth and Planetary Sciences, University of Chinese Academy of Sciences, Beijing 100049, China
Keywords:
Natural gas hydrates
Methane hydrate
Molecular simulations
Hydrate nucleation
Hydrate growth
Hydrate formation
Nucleation theory
NGHs exploration trial engineering
Oil and gas exploration engineering
A B S T R A C T
How natural gas hydrates nucleate and grow is a crucial scientific question.The research on it will help solve practical problems encountered in hydrate accumulation, development, and utilization of hydrate related technology.Due to its limitations on both spatial and temporal dimensions, experiment cannot fully explain this issue on a micro-scale.With the development of computer technology, molecular simulation has been widely used in the study of hydrate formation because it can observe the nucleation and growth process of hydrates at the molecular level.This review will assess the recent progresses in molecular dynamics simulation of hydrate nucleation and growth, as well as the enlightening significance of these developments in hydrate applications.At the same time, combined with the problems encountered in recent hydrate trial mining and applications, some potential directions for molecular simulation in the research of hydrate nucleation and growth are proposed, and the future of molecular simulation research on hydrate nucleation and growth is prospected.
1.Introduction
Natural gas hydrates (NGHs) are crystalline compounds,stable at low temperature and high pressure conditions (e.g.,temperature <10 °C and pressure >7 MPa).These compounds are made up of hydrogen-boned water cages (host) and enclathrated guest molecules, the guest molecules can be methane, ethane, carbon dioxide, propane, etc (Sloan ED,2003; Sloan ED and Koh CA, 2008).As shown in Fig.1,water and guest molecules make up five basic cage types (512,51262, 51264, 435663, 51268), while three common structures occur in nature from the topologic stacking of the five cage types in space.The unit cell of structure I hydrate consists of two small 512cages and six slightly larger 51262cages, the unit cell of sII hydrate is composed of sixteen small 512cages and eight medium 51264cages, while sH unit cell is made up of three small 512cages, two small 435663cages and one large 51268cage.
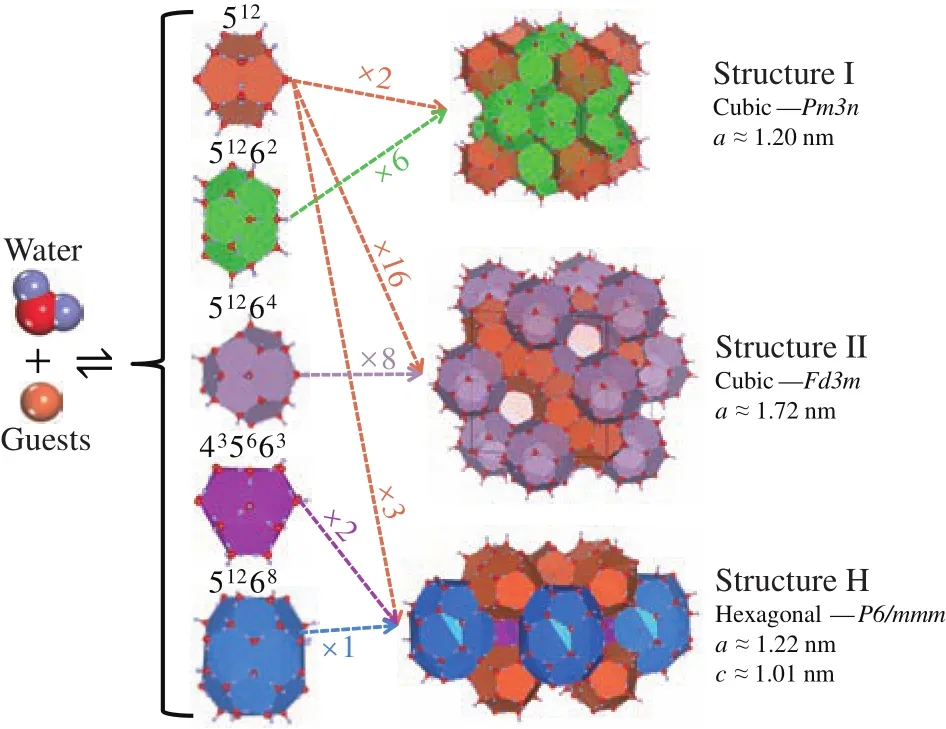
Fig.1.Cages and three common crystal structures of NGHs.Structures were generated with VESTA-3 (Momma K and Izumi F, 2011).
NGHs attract lots of attention because of their industrial and scientific importance.NGHs are regarded as potential energy resources because they are widespread in the permafrost and in continental-slope and -emergence ocean regions; they have approximately the same energy density as that of the compressed gas (e.g., methane molecules are separated ca.0.6 nm by the water cages, which is roughly same molecular distance as that of compressed gas at 273.15 K and 13 MPa); and the total organic carbon content of NGHs is almost twice that of all the other proved fossil fuels(Kvenvolden KA, 1993).Because of this, several countries,e.g., China, the USA, Japan, India, and Canada, plan to exploit NGHs, and China conducted two successful production tests in the South China Sea in 2017 and 2020 (Li JF et al., 2018; Qin XW et al., 2020; Ye JL et al., 2020).Besides their value as energy resources, NGHs can be used as media for storage and transportation of gases, mixed gas separation, and carbon dioxide sequestration, etc (Haugan PM and Drange H, 1992; Sloan ED, 2003; Koh CA et al., 2011;Hassanpouryouzband A et al., 2020; Zheng JJ et al., 2020).NGHs can also lead to undesirable side-effects on flow assurance (Sloan ED, 2003; Sum AK, 2013; Shi BH et al.,2021), e.g., gas hydrate formation might block offshore oilgas pipelines, causing huge economic and property losses.As a major component of NGHs and a greenhouse gas, methane released from NGHs decomposition from submarine might exacerbate the greenhouse effect (Kvenvolden KA, 1999;Andreassen K et al., 2017).In science, NGHs are crystal phases of water, and might widely exist in other planets of the universe (Loveday JS et al., 2001; Falenty A et al., 2014;Kamata S et al., 2019).
While the thermodynamic properties of NGHs have been studied extensively and well established (Sloan ED and Koh CA, 2008; Hester KC and Brewer PG, 2009; English NJ and MacElroy JMD, 2015), there are still lots of open questions related to the formation of NGHs (Ripmeester JA and Alavi S,2016; Guo GJ and Zhang ZC, 2021).One of the reasons for this is that it is quite challenging for the experiments to observe the nanoscale processes in both spatial and temporal dimensions.Molecular simulation, as an efficient tool to investigate microscopic structures and dynamics in physical or chemical processes, is playing an essential role in the research on NGHs formation.This can be revealed by Fig.2 that the number of publications in this area is rising in the last 15 years.
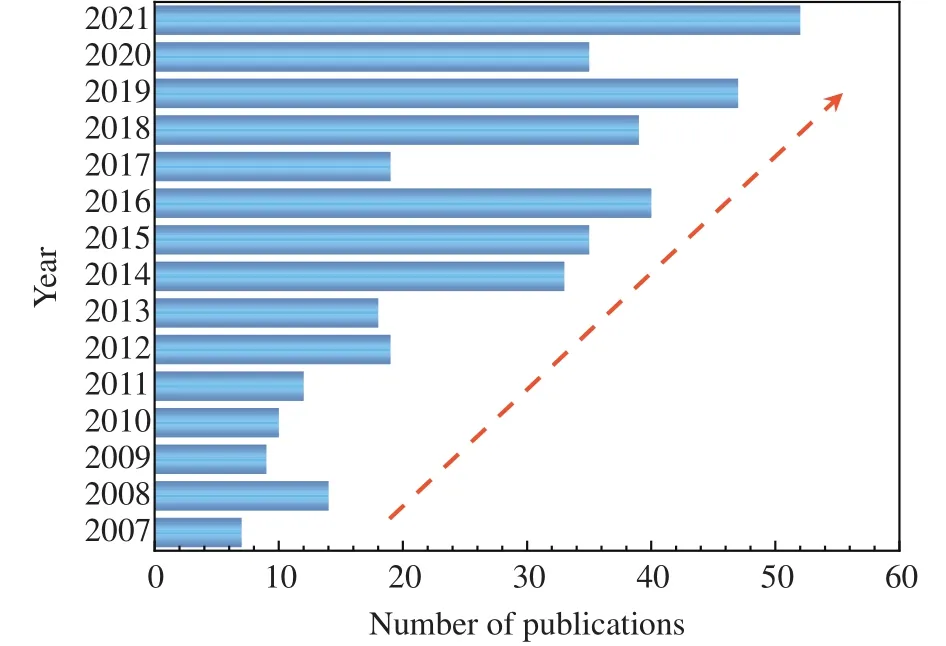
Fig.2.The number of publications from the past 15 years on molecular simulation studies of gas hydrate formation.Data is from the Web of Sciences with keywords of “gas hydrate formation” and“molecular simulation”.
Akin to the common crystal formation processes, NGHs formation involves the formation of an embryo, i.e.,nucleation, and the growth of the post-critical nucleus.As the nucleation of the NGHs embryo means a solid phase developing from liquid, which involves a random induction time and occurs at the nano- to micro-scale, therefore, it is a harder scientific problem than NGHs growth.In this paper,we will review recent advances in molecular simulation studies on NGHs nucleation and growth, especially on nucleation, as well as elusive challenges thereof.In addition,theoretical researches on gas hydrate formation in the future and potential applications of molecular simulations in hydrate exploitation are discussed.
2.Overview of NGHs formation studies
2.1.NGHs nucleation
The development of molecular simulation is inseparable from the development of computational technology.As a result, although the first reported molecular simulation study on NGHs nucleation was performed in 1997 (Hirai S et al.,1997), it is unphysical due to freezing pre-arranged guest molecules.Until 2009 the first simulation trajectory of spontaneous methane hydrate nucleation from water-methane phase was reported (Walsh MR et al., 2009).A brief history of studies on hydrate nucleation is indicated in Table 1.Before hydrate nucleation was observed by molecular dynamics (MD) simulations, several conceptual scerarios were proposed for hydrate nucleation.Sloan and Fleyfel proposed the labile cluster hypothesis (LCH) in 1991 (Sloan ED and Fleyfel F, 1991).As shown in Fig.3, in the initial condition, the system has reached target temperature and pressure conditions and there are some hydrogen-bonded water rings in it, but no gas molecules are present in the water; once gas molecules dissolve in the water, labile clusters form rapidly; these clusters either agglomerate by sharing faces or dissociate; once the size of the cluster reaches a critical value, hydrate growth commences.This hypothesis is based on experimental observations and lack of molecularlevel evidence because at that time, molecular simulation study on NGHs formation is unavailable.Afterwards, several experimental techniques were used to study NGHs formation(Ye YG et al., 2003), the strongest evidence supporting this hypothesis is that the hydration number of aqueous methane was determined to be 21 by magic-angle spinning (MAS)NMR spectrum (Dec SF et al., 2006), which is close to the number of water molecules in a 512cage (20).By using molecular simulations, Guo et al.found that the lifetime of single hydrate cages in liquid water presents a lognormal distribution, and a few cages have a quite long lifetime, which supports the LCH to a certain extent (Guo GJ et al., 2004,2005).
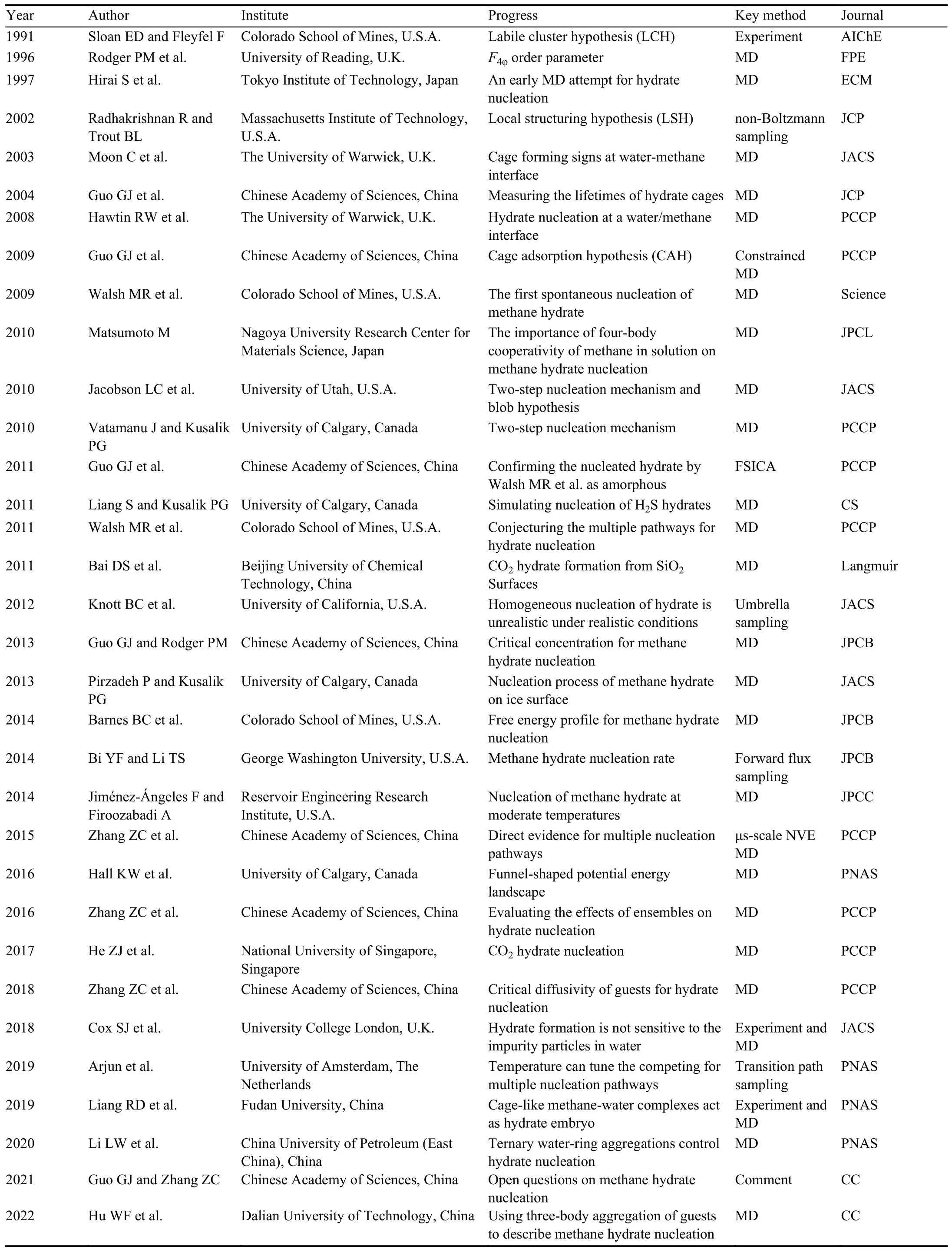
Table 1.A brief history of studies on gas hydrate nucleation, ordering with the publication date strictly.
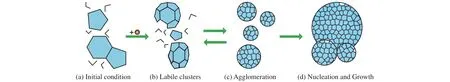
Fig.3.Schematic model of LCH proposed by Sloan and co-workers (after Sloan ED and Fleyfel F, 1991; Christiansen RL and Sloan ED,1994).
In 2002, Radhakrishnan and Trout proposed a local structuring hypothesis (LSH) based on Monte Carlo simulations and non-Boltzmann sampling of carbon dioxide hydrate nucleation (Radhakrishnan R and Trout BL, 2002).They proved that the LCH is not realistic because cages tend to separate in solution thermodynamically.For LSH, hydrate nucleation initiates in the following steps: guest molecules might stochastically arrange in a hydrate-like configuration which is caused by thermal fluctuations; once the number of guest molecules in this hydrate-like configuration exceeds the critical number, water molecules around methane molecules will relax and arrange to form cages structures, thus resulting in gas hydrate nucleation (see Fig.4).Rodger’s group performed simulations to study the structural evolution of a water film under a methane atmosphere (Moon C et al., 2003)and found that there is no evidence showing that the hydration shell of methane molecules resembles cage structures, but they observed the arrangement of methane molecules within the cluster.These results and their successive article (Hawtin RW et al., 2008) thus support the LSH.Guo et al.’s simulation results indicated that 512cages can adsorb dissolved methane, and their lifetime increases exponentially with the number of methane adsorbed, which can trigger selforganizing methane accumulation (Guo GJ et al., 2007), thus supporting the LSH.However, although the LSH points out that thermal fluctuations may induce the formation of local structuring guest molecules, the details are missing.

Fig.4.Schematic model showing the local structuring hypothesis(LSH).Six snapshots along nucleation are depicted.Carbon dioxide molecules are in cyan.White lines mean hydrogen bonds between water molecules.Modified with permission from Radhakrishnan R and Trout BL (2002).
The thermodynamic driving force for the local arrangement of guest molecules deserves to be explored.For this purpose, Guo et al.calculated the potential mean force between hydrate cages and guest molecules in water (Guo GJ et al., 2009).An adsorption interaction (stronger than hydrogen bonds) between hydrate cages and methane molecules was observed (Fig.5), therefore, Guo et al.proposed cage adsorption hypothesis (CAH) for NGHs nucleation.The hydrate cages (either standard or nonstandard)spontaneously form in methane aqueous solution, and these cages can adsorb surrounding methane molecules; the water molecules around the adsorbed methane arranges to form more hydrate cages; when the size of cage cluster is large enough, hydrate grows inevitably.CAH conjectures that the formed hydrate can be either crystalline or amorphous hydrate because the nucleus can be made up of either standard or nonstandard hydrate cages (Guo GJ et al., 2009).CAH can also predict the critical concentration for methane hydrate nucleation being 0.044 mol fraction, where the average separation between methane molecules is about 0.88 nm,locating the first peak of cage-methane PMF (Guo GJ and Rodger PM, 2013; Guo GJ and Zhang ZC, 2021).
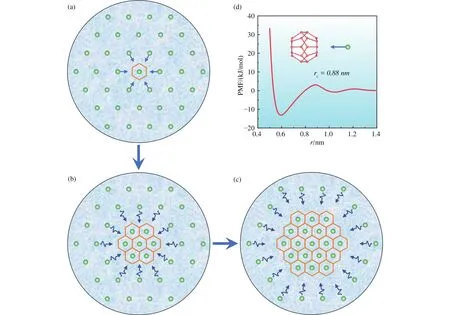
Fig.5.The cage adsorption hypothesis (CAH).When the guest concentration reaches the critical value (for methane, corresponding to the average separation between them being 0.88 nm), the first spontaneous formed cage will adsorb the neighboring guests (a), then water molecules will rearrange to form more cages and more guests will diffuse toward the cage cluster (b).This process will go on and when the size of cage cluster is large enough, hydrate grows inevitably (c).The potential mean force (PMF) between 512 cage and methane molecule (d).
In 2009, Walsh et al.conducted microsecond simulations with a water/gas two-phase system (see Fig.6a) and first realized the spontaneous nucleation of methane hydrate using molecular dynamics simulations (Walsh MR et al., 2009).The resulting hydrates were further examined to be amorphous by using the face-saturated incomplete cage analysis (FSICA)program (Guo GJ et al., 2011).Walsh et al.’s work is a milestone that makes researchers believe that hydrate nucleation processes can be observable by using molecular simulations, thus massive molecular simulation studies were performed on hydrate nucleation of various guest molecules,such as methane (Jacobson LC et al., 2010a, 2010b;Vatamanu J and Kusalik PG, 2010; Walsh MR et al., 2011a;Walsh MR et al., 2011b; Sarupria S and Debenedetti PG,2012; Guo GJ and Rodger PM, 2013; Bi YF and Li TS, 2014;Jime?nez-A?ngeles F and Firoozabadi A, 2014; Lauricella M et al., 2015; Yuhara D et al., 2015; Zhang ZC et al., 2015, 2016),carbon dioxide (Bai DS et al., 2011, 2012, 2015a, 2015b; He ZJ et al., 2016, 2017b), hydrogen sulfide (Liang S and Kusalik PG, 2010, 2011a, 2011b, 2013, 2015a), ethane(Wilson DT et al., 2016), propane (Jime?nez-A?ngeles F and Firoozabadi A, 2015), and THF (Wu JY et al., 2016).
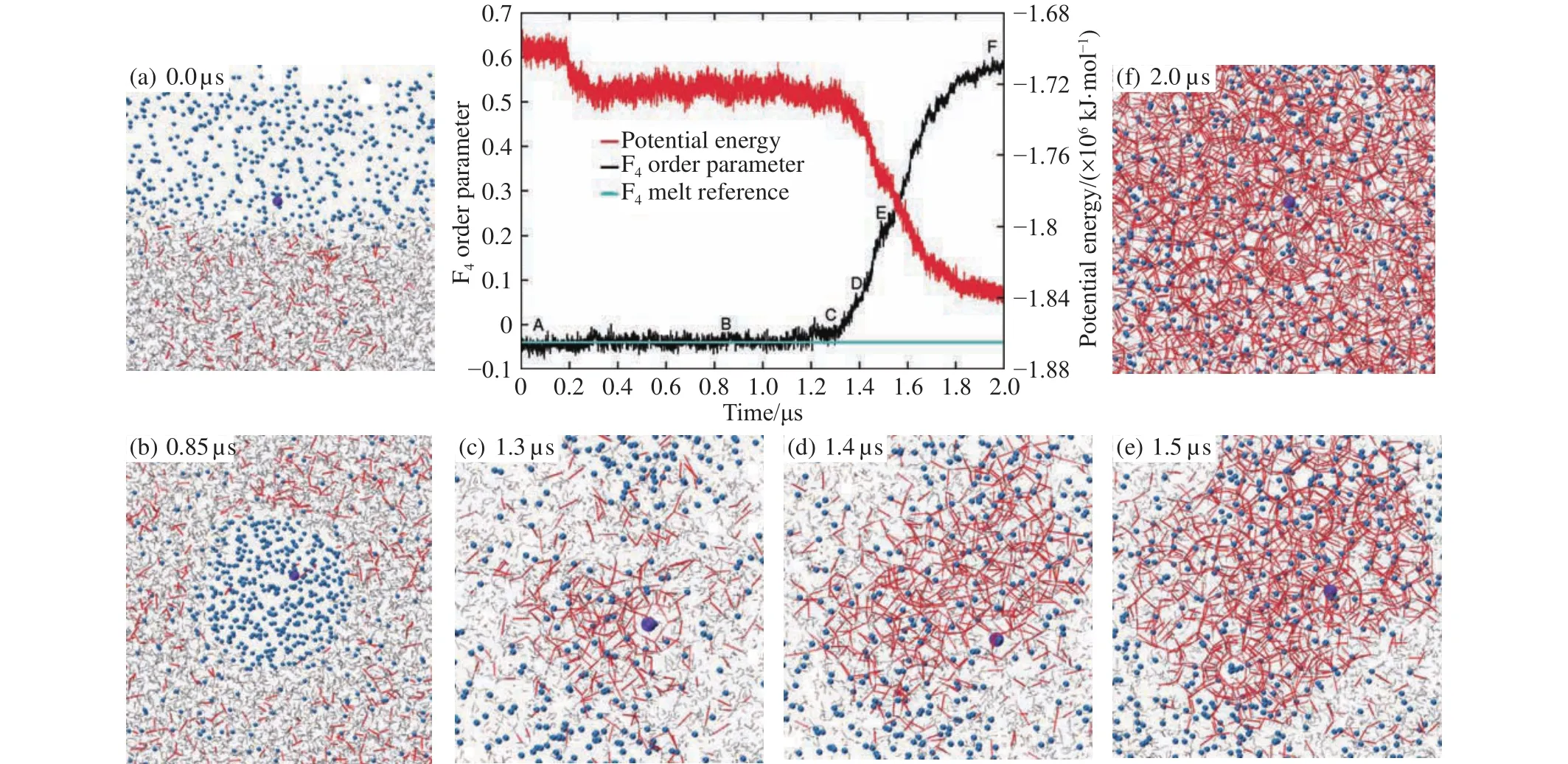
Fig.6.Snapshots, potential energy, and order parameter showing the first spontaneous formation trajectory of methane hydrate from microsecond molecular simulations at 250 K and 50 MPa.Methane is represented as blue spheres, and hydrogen bonds between water molecules in red lines.The figure reproduced with permission from Walsh MR et al.(2009).
As the molecular level details of the hydrate nucleation process have been revealed by molecular dynamics simulations, the nucleation mechanism was figured out irresistibly.The two-step nucleation mechanism for gas hydrate was proposed by Molinero ’s group and Kusalik ’s group almost simultaneously, where hydrate nucleation proceeds with the first step of forming amorphous clusters,and then these amorphous hydrate transforms into crystalline hydrate (Jacobson LC et al., 2010b; Vatamanu J and Kusalik PG, 2010).Molinero’s group also pointed out that the amorphous clathrate was born from a blob consisting of concentrated guests in water-mediated configurations (see Fig.7).In 2020, Li et al.proposed that the ternary water-ring aggregations (TWRAs) is the fundamental structures for gas hydrate nucleation and a methane hydration layer compression/shedding process is the driving force for the formation of the blob, which was regarded as the critical step for hydrate nucleation (Li LW et al., 2020).Further, Hu WF et al.proposed that the three-body aggregation of methane is also a key step in hydrate nucleation but from another perspective of guest molecules (Hu WF et al., 2022).However, it is still questionable that whether the metastable amorphous phase is indispensable.
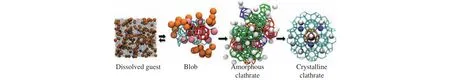
Fig.7.Schematic for the two-step mechanism as proposed by Molinero’s group.The figure is reproduced with permission from Jacobson LC et al.(2010b).
The above-mentioned question was answered by later simulation studies.Walsh et al.found that some of the formed amorphous hydrates from their simulations show long-range order, thus conjecturing that the system might directly nucleate to crystalline phase (Walsh MR et al., 2011b).This was proved by the microcanonical molecular simulations of methane hydrate nucleation as shown in Fig.8 and the multiple pathways for hydrate nucleation were illustrated(Zhang ZC et al., 2015).Recent atomic insight into the multiple pathways revealed that there are competing mechanisms between two-step nucleation and direct nucleation for methane hydrate, while with increasing temperature, the mechanism shifts from two-step to direct crystalline hydrate nucleation (Arjun et al., 2019).
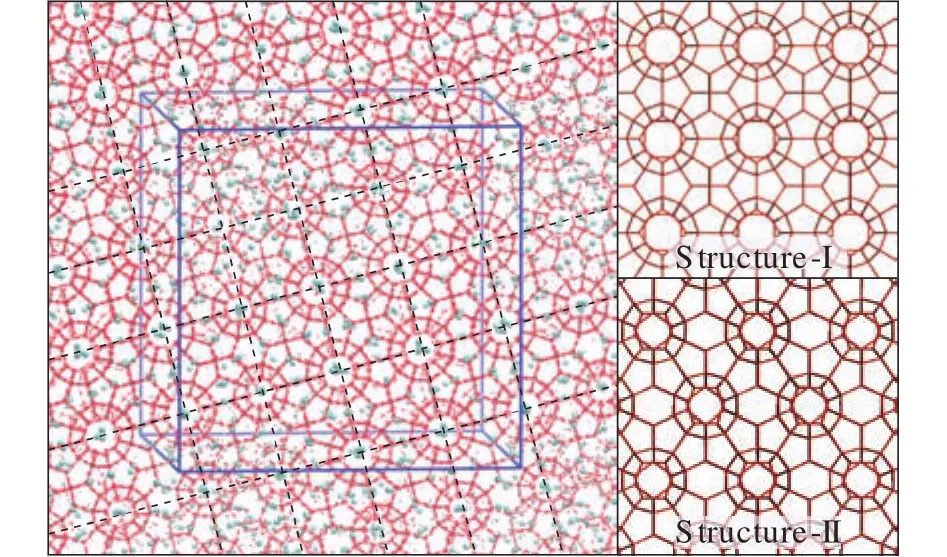
Fig.8.Snapshots from one of the nucleation trajectories of methane hydrate, where direct nucleation of crystalline hydrate was observed.The figure was reproduced with permission from Zhang ZC et al.(2015).
While hydrate nucleation mechanisms were investigated by numerous molecular dynamics simulation studies, how to control hydrate nucleation was deemed the next question.Guest concentration in solution was regarded as the most important in the first place, for example, hydrate nucleation can be achieved in high concentration gas solution even at high temperatures (e.g., 285 K) (Jime?nez-A?ngeles F and Firoozabadi A, 2014, 2015).This phenomenon was explained by Guo and Rodger that there was a critical concentration for hydrate nucleation, and if the concentration is greater than this, hydrate nucleation occurs (Guo GJ and Rodger PM,2013).However, a subsequent study predicted that methane and carbon dioxide hydrate nucleation would have similar critical concentrations according to the similar locations of the first peak in cage-guest PMF (Liu CJ et al., 2016), which was inconsistent with the simulation results, that is, ~0.04 for methane (Zhang ZC et al., 2016) but ~0.09 for carbon dioxide(He ZJ et al., 2017b).In this scenario, research on common characteristics of hydrate nucleation from different guest molecules turns to be crucial.Hence, the properties of aqueous solutions of seven guest molecules (methane, carbon dioxide, argon, krypton, xenon, radon, and propane) and their relationship with nucleation of hydrate were studied by molecular dynamics simulations (Zhang ZC et al., 2018a).The results show that with increasing solution concentration,the hydration shell of guest molecules becomes more and more ordered, and the total entropy of the system decreases.These changes increase the free energy of gas solution, which is conducive for the system to cross the energy barrier and trigger the nucleation of hydrate (Fig.9b).Interestingly, there is a common critical self-diffusivity of guest molecules at each temperature, at the critical point, different guests have different concentrations but common diffusivity.Hydrate quickly nucleates as long as the self-diffusivity of guest molecule is lower than the critical value (see Fig.9).It emphasizes the crucial role of guest dynamics on hydrate nucleation.In Zhang ZC et al.'s following works, the larger hydrocarbon molecules, e.g., ethane, propane, and 2.2-dimethylbutane, were examined to be effective promoters for methane hydrate nucleation because of their slower dynamics than methane's in solution (Zhang ZC et al., 2019, 2020).
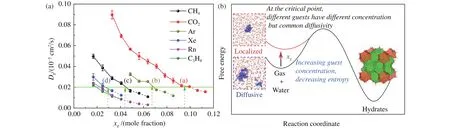
Fig.9.(a) Critical self-diffusivity for gas hydrate nucleation and (b) schematic for thermodynamic of hydrate nucleation.Figures reprinted with permission from Zhang ZC et al.(2018a).
The thermodynamic and kinetic properties of hydrate nucleation were also evaluated through molecular simulations since 2009.The nucleation rate of gas hydrate was estimated based on direct simulations (Walsh MR et al., 2011a),umbrella sampling (Knott BC et al., 2012), equilibrium path sampling (Barnes BC et al., 2014), forward flux sampling (Bi YF and Li TS, 2014), mean-first passage time (Yuhara D et al., 2015; Zhang ZC et al., 2016; Lauricella M et al., 2017),survival probability (Yuhara D et al., 2015; Lauricella M et al., 2017), meta-dynamics (Lauricella M et al., 2014), and transition path sampling (Arjun A and Bolhuis PG, 2020,2021).As for the thermodynamic properties, forward flux sampling & backward flux sampling (Bi YF et al., 2016),equilibrium path sampling (Barnes BC et al., 2014), and transition path sampling (Arjun et al., 2019) were used to assess the free energy landscape of hydrate nucleation, the authors claimed that the free energy profile can be well fitted with the classical nucleation theory despite that there are noticeable differences for the small nucleus (Barnes BC et al.,2014; Bi YF et al., 2016).
Although the above-mentioned works are all on homogeneous nucleation processes, Knott et al.’s simulations argued that heterogeneous nucleation is the only accessible mechanism in nature for methane hydrate (Knott BC et al.,2012).Bai et al.realized heterogeneous nucleation of carbon dioxide hydrate on the solid surface of silica and found that solid layer and three-phase contact region were conducive to nucleation, respectively (Bai D et al., 2011, 2012, 2015a).Liang et al.found that methane hydrate can nucleate on the surface of silica (Liang S and Kusalik PG, 2015a).Yan et al.found that the pore size of clay minerals has a great influence on the nucleation process of methane hydrate (Yan KF et al.,2016).He et al.found that the hydrophilic (silica) and hydrophobic (graphite) surfaces affect the concentration of methane or carbon dioxide in the solution phase, thus affecting the nucleation process (He ZJ et al., 2017a; He ZJ et al., 2021).Pirzadeh and Kusalik reported the nucleation process of methane hydrate on ice surface (Pirzadeh P and Kusalik PG, 2013).Our previous simulations indicated that methane hydrate can nucleate heterogeneous or homogeneous in the quasi-liquid layer of ice surface (Zhang ZC and Guo GJ, 2017).Experiments and simulations from Cox et al.showed that methane hydrate formation is not sensitive to the impurity particles in water (Cox SJ et al., 2018).In summary,it is still under debate from all the reported simulation works in the presence of foreign particles or minerals on gas hydrate nucleation — whether hydrate nucleation prefers the foreign surfaces, in other words, whether heterogeneous nucleation is the preferred mechanism.In addition, nucleation of hydrate in the confined space is a more complicated problem, which deserves to be evaluated.
2.2.NGHs growth
Compared to hydrate nucleation, NGHs growth is easy to be realized by molecular simulations because there is no induction time for hydrate growth.The first topic on hydrate growth is the growth mechanism.Kusalik’s group revealed that sI hydrate can interconvert to sII hydrate with the help of non-standard hydrate cage 51263(Vatamanu J and Kusalik PG,2006a, 2006b, 2008), sI hydrate can cross-nucleate sH hydrate with the help of 4151062cage, and sII hydrate can directly connect sH hydrate (Liang S and Kusalik PG, 2015b) (see Fig.10).
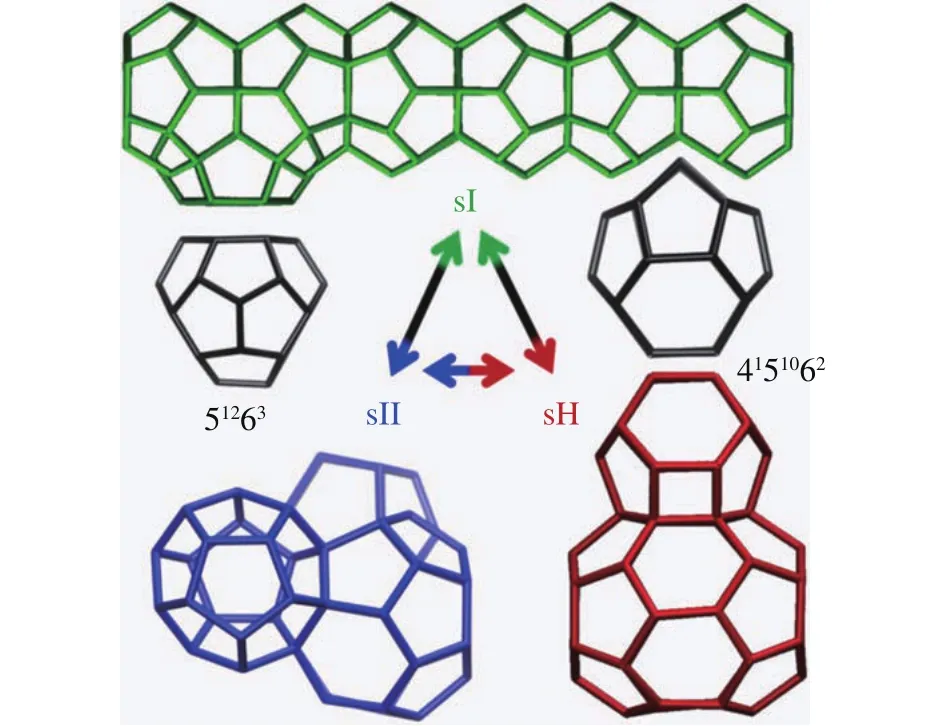
Fig.10.Possible structural interconversion between principal gas hydrate structures sI, sII, and sH.Reprinted with permission from Liang S and Kusalik PG (2015b).
Then, factors affecting hydrate growth were assessed by molecular simulations.It was reported that the growth rate of methane hydrate is dominated by the concentration of methane in solution, the diffusivity of methane in water, and the adsorption of methane by the hydrate-solution interface(Tung Y-T et al., 2010, 2011, 2012).Focused on these factors,it was found that the attractive force between hydrate surface and guest molecules is sub-nanoscale, it implies that this force does not affect the mass transfer of gas hydrate growth(Yagasaki T et al., 2015), therefore solubility and diffusivity of guest molecules dominate the growth process (Zhang ZC et al., 2018b).Further simulation works showed that the temperature dependence of hydrate growth rate has a maximum value as shown in Fig.11 (Yagasaki T et al.,2016a; Zhang ZC et al., 2018b; Tian HQ and Zhang ZC,2020), similar to that of ice growth (Rozmanov D and Kusalik PG, 2011), which is the synergy effect of the thermodynamics and kinetics of molecules (e.g.for hydrate, the increasing temperature lowers the concentration of guest molecules in solution but increases the kinetics of molecules at the interface).As for the growth of sII hydrate, the larger guest molecule might be trapped in the open small cage at the hydrate surface, which will hinder hydrate growth (Wu J-Y et al., 2016; Yagasaki T et al., 2016a).Besides above-mentioned factors, heat and mass transfer processed might impact hydrate growth, however, they were rarely studied.
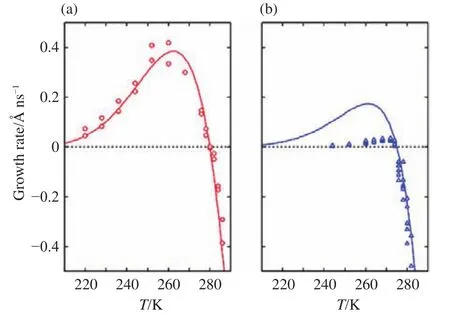
Fig.11.Growth rates of (a) ethylene oxide hydrate and (b) THF hydrate.Reprinted with permission from Yagasaki T et al.(2016b).
Inhibition of hydrate growth is one of the effective methods to prevent hydrate formation, while molecular simulation is a powerful tool to examine the performance of a variety of hydrate growth inhibitors.As for thermodynamic inhibitors, such as NaCl and methanol, they can lower the melting temperature of gas hydrate and induce the formation of gas bubble from the saturated guest solution generated by hydrate decomposition, thus efficiently inhibiting hydrate growth (Yagasaki T et al., 2014a, b).However, they are dosed at 20%?50%, which is uneconomical, in comparison with low-dosage inhibitors (Koh CA et al., 2011).For low-dosage hydrate inhibitors, they are two kinds, one of them is kinetic hydrate inhibitors (KHIs) and another is the anti-agglomerants(AAs).KHIs inhibit hydrate growth by adsorption on the hydrate surface, which will create a curved hydrate interface,thus preventing or hindering the further growth of hydrate.Therefore, the key factors affecting the performance of KHIs are the size of KHIs, the adsorption strength between hydrate surface and KHIs, and the relaxation time of KHIs on the hydrate surface.Focused on these factors, several simulation attempts were made to develop better KHIs (Yagasaki T et al.,2015; Xu JF et al., 2018; Yagasaki T et al., 2018a, 2018b).AAs inhibit hydrate formation by preventing the agglomeration of small hydrate particles, where AAs were adsorbed on the hydrate surface, which will avoid the contact of hydrate particles.The adsorption of AAs on hydrate surface, the effects of additives on the performance of AAs,and the effect of AAs on hydrate growth have been investigated by molecular simulations (Bellucci MA et al.,2018; Bui T et al., 2018; Mehrabian H et al., 2018; Mehrabian H et al., 2019; Naullage PM et al., 2019; Bui T et al., 2020;Mehrabian H and Trout BL, 2020; Mohr S et al., 2021).
Promotion of the growth of hydrate is highly related to the gas storage in the form of gas hydrate, carbon dioxide sequestration in hydrate, etc.Surface surfactant, soluble larger molecules were proposed and examined as effective promoters for gas hydrate by using molecular dynamics simulations (Ji HQ et al., 2017; Lin ZY et al., 2018; Zhang ZC et al., 2019; Wang PW et al., 2021).
3.Challenges/knowledge gaps/needs
3.1.NGHs research
In the past decades, researchers have made great progress on the formation mechanism of NGHs.Since the hypothesis of hydrate nucleation was put forward, the understanding of the hydrate formation mechanism has been continuously deepened.In recent years, the development of science and technology has allowed us to qualitatively describe the formation mechanism of hydrates at the molecular level.Therefore, according to the law of scientific development,scholars have qualitatively described the hydrate nucleation process of a single guest, multi-component guest, and multiphase coexisting systems.However, some questions related to the formation of NGHs are still unresolved, which is reflected in the fact that people cannot quantitatively predict and precisely control the formation process of hydrate.There is still a lack of mature theory to guide the development of hydrate-related technologies, and unexpected results are often obtained, which is unpredictable, highlighting the lack of support from the basic theory.
Classical nucleation theory (CNT) was regarded as the most successful one during the past century to describe the nucleation process, including gas to liquid and liquid to solid,however, CNT cannot predict the formation of hydrate, which is mainly manifested in the following aspects: (1) most hydrate nuclei are amorphous and not the most stable crystalline phase assumed by CNT (Sosso GC et al., 2016);(2) hydrate nucleation may require two or more steps which do not meet the assumption of CNT (Jacobson LC et al.,2010b; Vatamanu J and Kusalik PG, 2010; Arjun et al.,2019); (3) the hydrate nucleus is unnecessarily spherical,which does not meet the CNT assumption (Barnes BC et al.,2014; Bi YF et al., 2016); (4) the hydrate nucleus is multicomponent, and the ratio of the components and the structure of the nucleus all influence the stability of the nucleus, thus impacting the size of the critical nucleus, which is not considered in the CNT.In this scenario, it takes a long way to get a predictive theory of hydrate nucleation.
3.2.Molecular dynamics simulation method
Theoretically, molecular simulation can realize the simulation of any physical or chemical process, but its application is currently very limited.As far as the simulation scale is concerned, we can only simulate at the microsecond and nanometer at present, which is far from the macroscopic scale.This means that many processes from microscopic to macroscopic cannot be observed through molecular simulations.
At the same time, the current molecular simulation force fields are not perfect.They can only reproduce part of the properties of molecules or ions.For example, there are currently dozens of force fields for water molecules;TIP4P/Ice is widely used in hydrate studies because it can reproduce the freezing point of water and the phase state of hydrate very well (Abascal JLF et al., 2005; Michalis VK et al., 2015), but in terms of the condensed phases of water molecules, TIP4P/2005 is better (Abascal JLF and Vega C,2005).At present, machine learning has been applied in molecular simulation, and it is expected that better molecular simulation force fields can be obtained through machine learning methods (Chan H et al., 2019; Mueller T et al.,2020).
In the gas hydrate formation experiment, system temperature and pressure are generally controlled by the water bath and pressure of the gas phase.Temperature or pressure jump can be monitored as the start of hydrate formation.As for simulations, thermostat and barostat algorithms are used to control the system conditions, which is quite different from that of experiments because these algorithms nearly immediately remove the exothermic heat of formation on nucleation and during growth.Microcanonical molecular simulation is regarded as an alternative for counting the heat transfer in the system (Liang S and Kusalik PG, 2013; Zhang ZC et al., 2015), nevertheless, the pressure cannot be controlled at the same time.Moreover, because the system size is not large enough, the temperature of the system will become too high.Therefore, simulating the experimental process of hydrate formation by molecular dynamics is still challenging.
4.Future research guidelines
4.1.Theoretical research on gas hydrate formation in the future
Current knowledge on gas hydrate nucleation indicates that CNT is not adequate for describing the nucleation processes.Thus, a mature theory on gas hydrate formation is highly required.Previous results suggest that the ordering from liquid to crystal involves several stages as that of Fig.12a, where order parameter dependence of energy is shown.Regarding the configurational space, the landscape is rugged(see Fig.12b), where the system needs to find the path from the liquid to the crystal and if the system firstly samples the glass state, it might be trapped in the amorphous state (local minima).Combing the characteristic feature of rugged configurational landscape and evolution of potential energy, a funnel-shaped potential energy landscape was proposed (Hall KW et al., 2016), where the system’s potential energy dictates the depth of the point in the funnel, and the funnel’s width at a point is the system’s entropy.In this scenario, the system experiences an entropy reduction prior to the lower of energy in the nucleation process, thus resulting in a free energy barrier according to the equationΔG=ΔH?TΔS, which has been proved by previous water and gas hydrate nucleation studies (Pirzadeh P et al., 2012; Liang S et al., 2019).In addition, bearing in mind the recent reported key role of guest dynamics on hydrate nucleation (Zhang ZC et al., 2018a), an alternative route to generate hydrate nucleation theory based on the funnel-shaped energy landscapes and roles of guest dynamics might be promising.There are several open questions related to hydrate nucleation besides the current knowledge (Fig.13), which can be referred to in our comments (Guo GJ and Zhang ZC.2021).
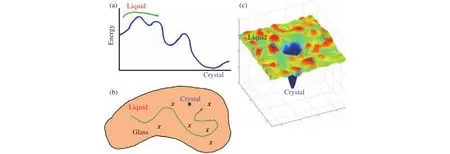
Fig.12.Schematic of the energy landscape for hydrate nucleation.The system’s potential energy dictates the depth of the point in the funnel,and the funnel’s width at a point is the system’s entropy in (c).Reprinted with permission from Liang S et al.(2019).
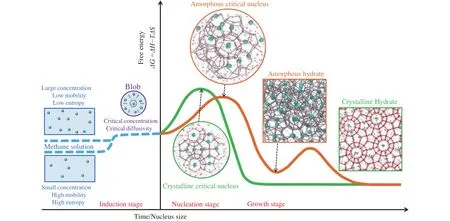
Fig.13.Schematic of methane hydrate nucleation.Reprinted with permission from Guo GJ and Zhang ZC (2021).
In addition, the amorphous phase was proved as the intermediate state of hydrate formation, however, how the amorphous hydrate transfers to the crystalline hydrate is still ambiguous.Chen et al.performed 20 μs long simulation under constant pressure and temperature ensemble to observe the evolution of hydrate structures nucleated from propane/methane/water system, nevertheless, no clear structure transition is observed (Chen Y et al., 2021).As previous studies reported cage transitions (Walsh MR et al.,2011b; Tang LL et al., 2012) and partial structural transition(Zhang ZC et al., 2015), a long simulation with larger system under microcanonical ensemble might be promising to resolve this problem.In addition, the evolution of grain boundaries of NGHs may act as sites for the initial transition of hydrate cages (Cao PQ et al., 2021; Zhang ZC et al., 2021), thus causing the transition from amorphous hydrate to crystalline hydrate.
4.2.Potential applications of molecular simulations in hydrate exploitation
Research on NGHs is an excellent example of research driven by both science and technology.The advancement of molecular simulations on hydrate formation definitely will be valuable in hydrate exploitation.In return, hydrate exploitation, especially, the trial mining of NGHs brings more questions that deserved to be explored in the future.The first one is where hydrate forms at first in submarine sediments?This is an important question related to how many hydrates may exist in the sediment.To answer this question, the effect of the surfaces of submarine sediment minerals, e.g., clay,quartz, and calcium carbonate, on hydrate formation needs to be evaluated.Then, the impacts of mineral pores on hydrate formation, including the size of pores and mineral components, should be assessed.Moreover, the research on the growth of hydrates in sediment environment may enlighten us why NGHs exhibits two types of occurrences in the pore’s spaces, i.e., pore-filling occurrence, and graindisplacing occurrence (Tréhu AM et al., 2006; Lei L and Seol Y, 2019; You K et al., 2019; Zhang YC et al., 2021a, 2021b).
The secondary formation of hydrate may block the well during exploitation (Li JF et al., 2018; Yang MJ et al., 2019;Qin XW et al., 2020; Wu NY et al., 2021).To inhibit hydrate formation, various attempts are proposed.One of them is to use hydrate inhibitors in drilling fluid to prevent hydrate formation.The second is to add modified nanoparticles into drilling fluid, which can both prevent the invasion of drilling fluid into NGHs deposits and prevent hydrate formation(Wang R et al., 2018; Guo DD et al., 2019).Apparently,molecular simulations as effective tools for hydrate formation research will be helpful on these problems.
5.Conclusion
Molecular dynamics simulations were used with success during the last two decades to understand the nucleation and growth of NGHs at the molecular level.This review summarizes the studying progress of gas hydrate nucleation and growth.From hypothesis to the molecular-level mechanisms, then to the evaluation of the performance of kinetic hydrate inhibitors and anti-agglomerants, molecular simulations have greatly deepened the understanding of gas hydrate formation.However, the current knowledge seems to indicate that the current theory is not capable of describing gas hydrate nucleation process quantitatively and satisfactorily.Therefore, a mature theory on gas hydrate nucleation is needed.Despite the drawbacks of molecular simulation techniques, it is still a useful, effective, and promising tool in the future to recognize the mechanism of NGHs formation in submarine sediments and to evaluate the performance of various hydrate inhibitors.These research results will not only have important application value for efficient exploitation of NGHs and flow assurance of oil and gas pipeline but also have important scientific significance for enriching and perfecting crystal nucleation theory.
CRediT authorship contribution statement
Zheng-cai Zhang drafted the manuscript.All authors revised the final manuscript.
Declaration of competing interest
The authors declare no conflicts of interest.
Acknowledgement
The authors are grateful to the Supercomputing Laboratory at IGGCAS and Center for High Performance Computing and System Simulation, Pilot National Laboratory for Marine Science and Technology (Qingdao) for the allocation of computing time.This work is jointly supported by Pilot National Laboratory for Marine Science and Technology (Qingdao), the IGGCAS (IGGCAS-201903 and SZJJ201901), and the Chinese Academy of Sciences (ZDBSLY-DQC003).

- China Geology的其它文章
- China achieved fruitful results in oil-shale gas-coalbed methane exploration and development in 2021
- Formation mechanism, experimental method, and property characterization of graindisplacing methane hydrates in marine sediment: A review
- Distributed optical fiber acoustic sensor for in situ monitoring of marine natural gas hydrates production for the first time in the Shenhu Area, China
- Experimental investigation of hydrate formation in water-dominated pipeline and its influential factors
- Stability analysis of seabed strata and casing structure during the natural gas hydrates exploitation by depressurization in horizontal wells in South China Sea
- Application of wellhead suction anchor technology in the second production test of natural gas hydrates in the South China Sea