
Hereditary polyposis syndromes remain a challenging disease entity:Old dilemmas and new insights
2023-03-17 07:45FrederiknnePachlerAnnaByrjalsenJohnsdalKarstensenAnneMarieJelsig
Frederik R?nne Pachler,Anna Byrjalsen,John Gásdal Karstensen,Anne Marie Jelsig
Frederik R?nne Pachler,John Gásdal Karstensen,Danish Polyposis Registry,Gastrounit,Copenhagen University Hospital - Amager and Hvidovre Hospital,Hvidovre 2650,Denmark
Anna Byrjalsen,Anne Marie Jelsig,Department of Clinical Genetics,University Hospital of Copenhagen,Rigshospitalet,Copenhagen 2100,Denmark
John Gásdal Karstensen,Department of Clinical Medicine,University of Copenhagen,Hvidovre 2650,Denmark
Abstract In this editorial we present an overview and insights of the management of hereditary polyposis syndromes.The primary focus was on familial adenomatous polyposis,juvenile polyposis syndrome and Peutz-Jegher syndrome.Genetic testing has become increasingly available and is easier than ever to integrate into clinical practice.Furthermore,several genes have been added to the expanding list of genes associated with hereditary polyposis syndromes,allowing for precise diagnostics and tailored follow-up.Endoscopic evaluation of patients with hereditary polyposis syndromes is paramount in the surveillance strategies.Current endoscopic procedures include both diagnostic procedures and surveillance as well as therapeutic interventions.Recommendations for endoscopic procedures in the upper and lower gastrointestinal canal were described.Surgery is still a key component in the management of patients with hereditary polyposis syndromes.The increased cancer risk in these patients often render prophylactic procedures or intended curative procedures in the case of cancer development.Surgical interventions in the upper and lower gastrointestinal canal were described with relevant considerations.Development of chemopreventive medications is ongoing.Few drugs have been investigated,including nonsteroidal anti-inflammatory drugs.It has been demonstrated that cyclooxygenase-2 inhibitors may lower the number of polyps.Other medications are currently under investigation,but none have,to date,consistently been able to prevent development of disease.
Key Words:Hereditary polyposis;Familial adenomatous polyposis;Juvenile polyposis syndrome;Peutz-Jegher syndrome
lNTRODUCTlON
It is estimated that gastrointestinal(GI)polyps develop in 40%-50% of the population,and risk factors include increasing age,male sex,smoking,and meat consumption[1-4].The management of one or a few polyps is in many cases straight forward.The polyp is removed,and the histopathology,localization,number,and size guide the need for endoscopic follow-up.However,when multiple or rare types of polyps are detected or when a polyp is detected in young patients,the clinical work-up is less straightforward.In those cases,it may be relevant to consider whether the patient has a hereditary polyposis syndrome.It is important to distinguish polyposis syndromes from spontaneous polyps,as individuals with polyposis syndromes often have a considerable risk of GI cancer.Further,these patients may also have an increased risk of extraintestinal cancer and sometimes other manifestations that may contribute to increased morbidity and mortality[5-7].Furthermore,first degree relatives may be at risk,as many syndromes are inherited through an autosomal dominant or recessive inheritance pattern.
Clear recommendations for suspecting a polyposis syndrome are difficult to decide upon.The syndromes present with considerable intra- and interfamilial variability,and many patients only have a few polyps and a negative family history.In this paper familial adenomatous polyposis(FAP),juvenile polyposis syndrome(JPS)and Peutz-Jegher syndrome(PJS)were described,as these are the most common.
GENETlC REVOLUTlON
Some of the polyposis syndromes have been known for over a century as clinical and hereditary entities(Figure 1).Knowledge of the underlying genetic cause of the syndromes increased when it became possible to investigate variants in the human genome.The first generation of sequencing methods,Sanger sequencing,was developed and commercialized in the 1970s and 1980s.This technique represented a revolution in genetic technology and made it possible to integrate genetic testing in clinical diagnostics[8,9].Some genes associated with polyposis were discovered in the 1990s and early 2000s,including adenomatous polyposis coli(APC)associated with FAP,mutYhomologue,STK11,type IA bone morphogenetic protein receptor(BMPR1A)and axis inhibition protein 2(Figure 1).However,Sanger sequencing is time-consuming and expensive,and it was not until the second(next)generation sequencing methods were developed that further polyposis genes were detected.Next generation sequencing is a form of parallel sequencing that was integrated in clinical practice around 2010.It facilitated fast and cheap sequencing of several genes simultaneously.Thus,genes such as polymeraseepsilon(POLE),polymerase delta 1(POLD1),andNTHL1have been added as causative of hereditary polyposis[10,11](Figure 1).
EXPANDlNG THE PHENOTYPE
The increased knowledge of genetic causes has revealed that several polyposis syndromes have a phenotypic overlap and that at least in the GI tract they mimic each other.Thus,patients with adenomatous polyposis do not always have FAP but may have other rarer syndromes including a polymerase proofreading-associated syndrome where pathogenic variants are detected in the exonuclease domain ofPOLEandPOLD1orNTHL1-related polyposis[12,13].Concerning hamartomatous polyposis syndrome,JPS is sometimes misdiagnosed because juvenile polyps are mistaken for inflammatory polyps[14].Furthermore,a mixture of polyps with different histopathology sometimes blurs the clinical picture,e.g.,in PTEN-hamartoma tumor syndrome(Cowden syndrome,Figure 2)where adenomas and inflammatory,hyperplastic and juvenile polyps can be present[15].Purely based on clinical manifestations it is often impossible to tell one polyposis syndrome from the other.
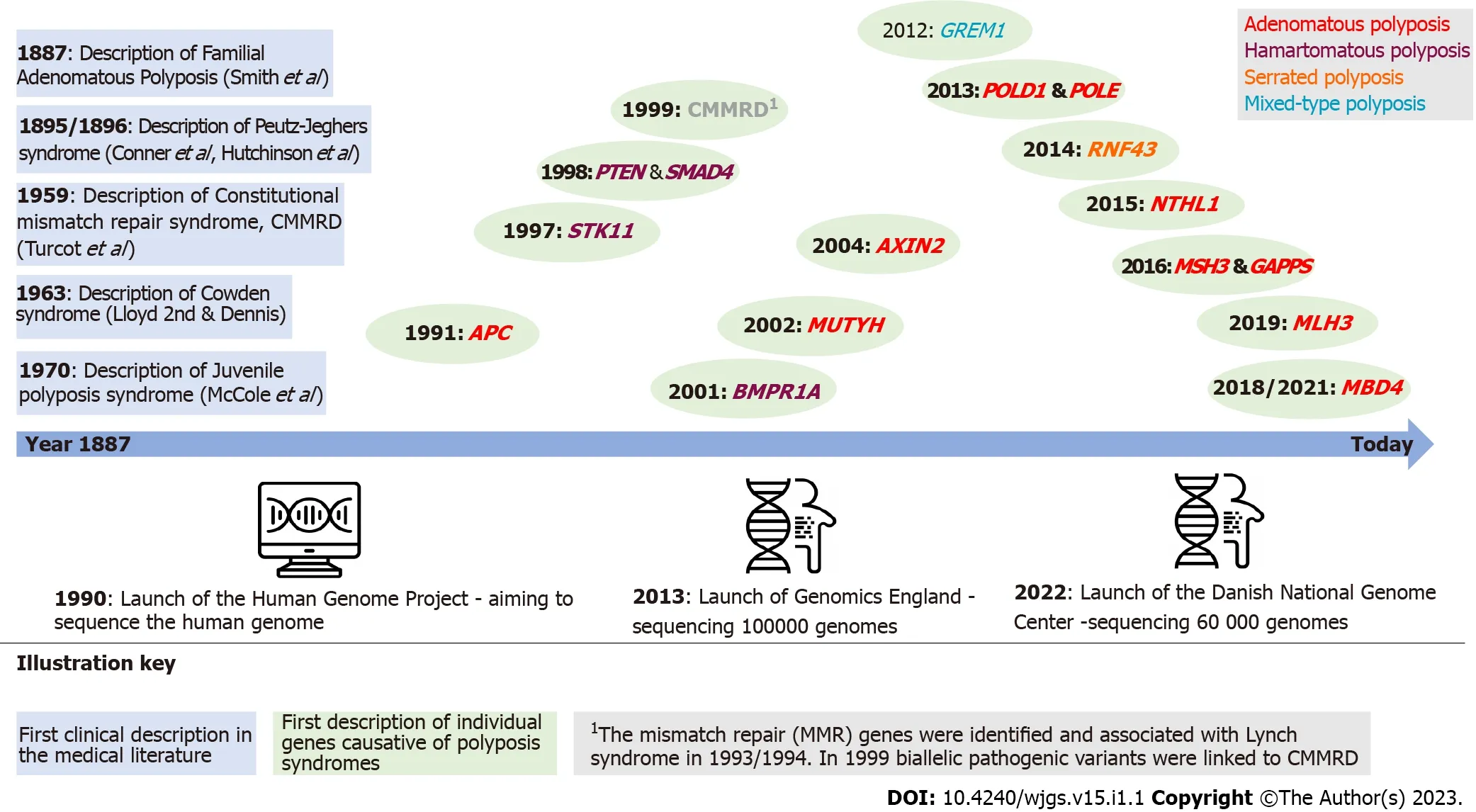
Figure 1 Timeline of hereditary polyposis syndromes and identification of causative gene.APC:Adenomatous polyposis coli;AXIN2:Axis inhibition protein 2;BMPR1A:Type IA bone morphogenetic protein receptor;CMMRD:Constitutional mismatch repair deficiency;POLD1:Polymerase delta 1;POLE:Polymerase-epsilon;MLH:MutL homolog;MUTYH:MutY homologue;RNF43:Ring finger 43.
It is important to know the genetic subtype,as the risk profile of the patient is different from syndrome to syndrome.The risk of extraintestinal cancer differs with the subtype,e.g.,a patient withNTHL1-related polyposis has in addition to the risk of colorectal cancer an increased risk of breast and uterine cancer(Table 1).Accordingly,the surveillance strategies should be tailored.Genetic analysis should be integrated in the diagnostic work-up and should comprise a gene panel with polyposisassociated genes as seen in Table 1.Genetic analysis should be carefully interpreted,and if a pathogenic variant is detected,genetic counseling is recommended.
Some efforts have been made to clarify a possible genotype-phenotype correlation especially in the most well-known syndromes like FAP and PJS[16,17].However,these studies are limited by the small number of patients.Furthermore,the underlying mechanisms that drive cancer development in several of the polyposis syndromes are largely unknown,especially in the hamartomatous polyposis syndromes.A hamartoma-adenoma-carcinoma sequence has been proposed but has yet to be confirmed[18].It is believed that cancer development in FAP follows the adenoma-carcinoma sequence caused by dysregulation of the Wnt/β-catenin pathway[19].However,more knowledge on the details of the pathophysiology is necessary to understand the background for polyposis and(extra)intestinal cancer.
SURGlCAL MANAGEMENT OF HEREDlTARY POLYPOSlS
When a patient is diagnosed or suspected of having a hereditary polyposis syndrome,the primary management include an endoscopic baseline examination with histopathological evaluation of polyps as well as a physical examination with focus on extraintestinal manifestations as seen in the polyposis syndrome[20](Table 1).There is a phenotypic overlap,as polyposis for most of the syndromes(PJS excluded)is primarily located in the large intestine.SMAD4-related JPS and FAP often have polyposis in the upper GI tract(gastric and duodenal polyps,respectively).It is common for all syndromes that when endoscopic management is no longer sufficient then surgical resection is the treatment of choice.Several guidelines have been published on recommended surveillance.However,the evidence level is often low due to a limited number of patients and lack of long-term follow-up studies evaluating different surveillance protocols[20-24].
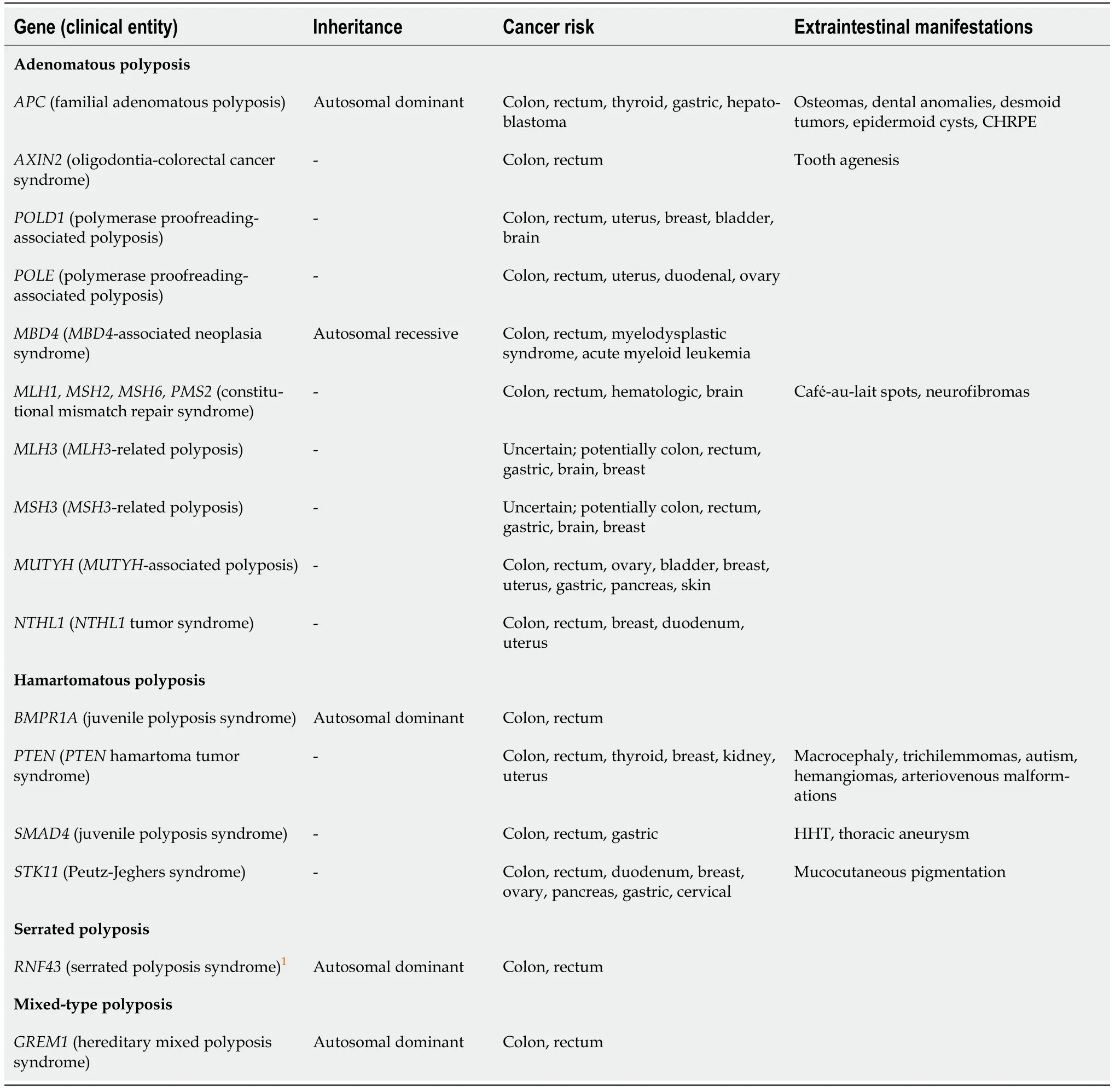
Table 1 Hereditary polyposis syndromes
LOWER Gl ENDOSCOPY AND SURGERY
Virtually all patients with polyposis syndromes undergo colonoscopy surveillance.The appropriate intervals and recommended age to initiate surveillance are debated.It is widely recommended that individuals with FAP are examined annually or biannually from the early teenage years[20,24].Development of cancer in patients with FAP is extremely rare before the age of 15[20,24-26],and it could be argued that endoscopic examinations should not start earlier than this.
During colonoscopy investigation,the use of chromoendoscopy,either direct or virtual with narrow band imaging,is often used.Narrow band imaging has not been shown to increase detection of neoplastic polyps,but it is a helpful tool in skilled hands[27].In patients with FAP a prophylactic colectomy is often indicated as most FAP patients will develop adenomatous lesions(Figure 2),and endoscopic surveillance is insufficient[20,24,26].However,it is now clear that patients who have a pathogenic variant inAPChave a very variable phenotype.Some may develop polyps at a later age,and thus family history should always be considered when recommending surveillance and deciding on surgery.Subtotal colectomy with ileorectal anastomosis(IRA)or proctocolectomy,often with the intent of restorative proctocolectomy with ileal-pouch anal anastomosis(IPAA),are recommended for patients with FAP[20,23,24,26,28].Surgery for patients who are known to have FAP from childhood often occur in their late teenage years but may be sooner or later if endoscopic findings dictate it.Surgery before the age of 15 years is not recommended[20,23,24,26,28].
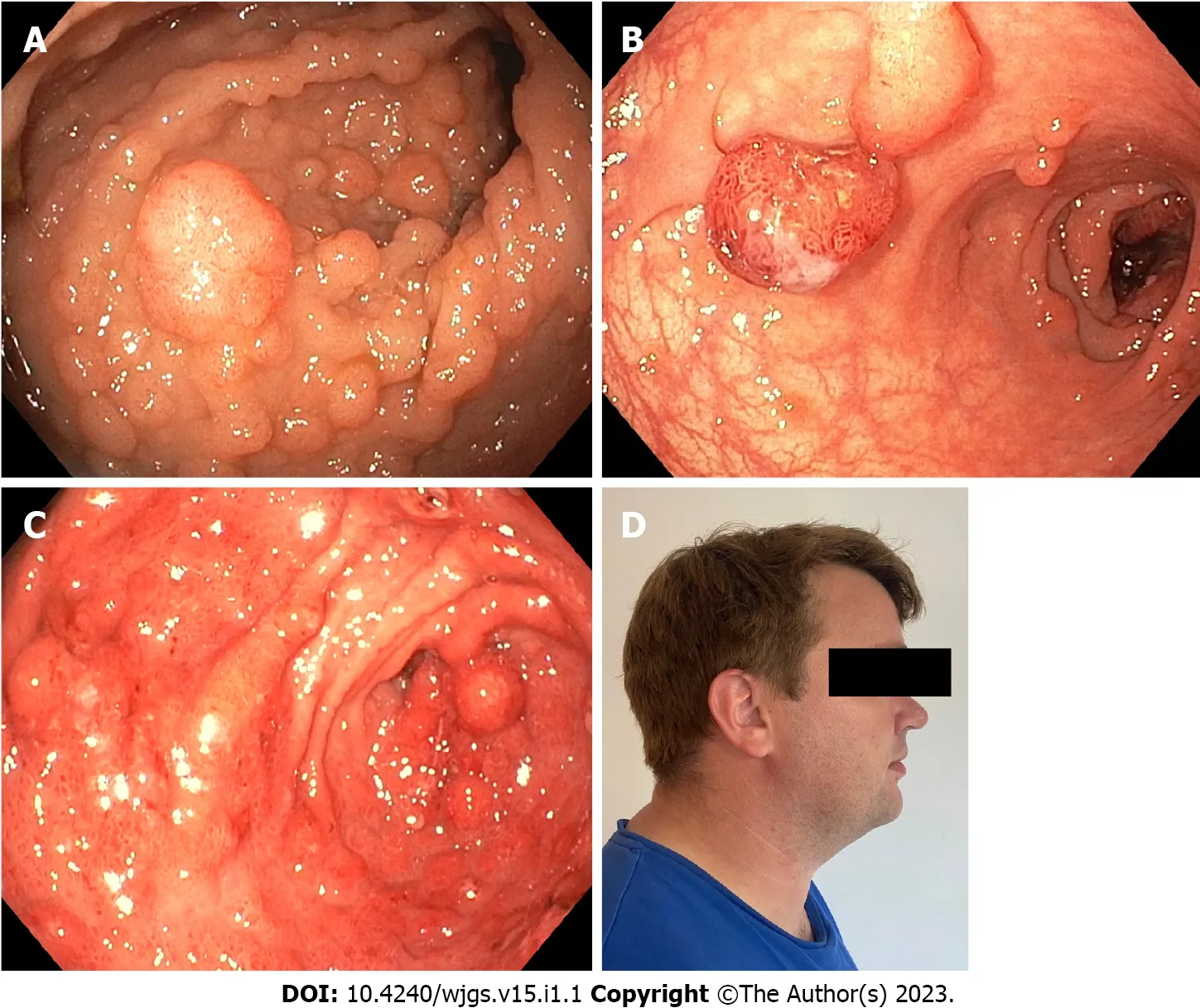
Figure 2 Polyps and extraintestinal manifestations in patients with hereditary polyposis syndromes.A:Severe colonic adenomatosis in a patient with familial adenomatous polyposis;B:Colonic polyposis in patient with Peutz-Jegher syndrome;C:Severe gastric polyposis in patient with SMAD4-related juvenile polyposis syndrome;D:Patient with Cowden syndrome and macrocephaly.
After colectomy endoscopic surveillance annually or biannually is recommended[20,24].However,it is heavily debated whether patients with IRA and patients with restorative proctocolectomy with IPAA should follow the same intervals.If polyposis progresses during surveillance in patients with IRA,proctectomy,possibly with IPAA,should be considered.This may be the appropriate choice,especially in patients with FAP,in the presence of numerous rectal polyps since excessive resection may lead to functional problems and technical difficulty in future proctectomy[20-22,28,29].
In all cases of colectomy,proctocolectomy with terminal ileostomy is also an appropriate solution.However,most patients undergoing resection are relatively young,and a permanent stoma may impact their quality of life[30].Hence,it is often desirable to attempt anastomosis with preserved continence,but this should be a subject for discussion and individualization.
In patients with JPS and PJS the initial colonoscopy is usually recommended around the age of 12 years for JPS[20,22,31]and 8-10 years for PJS[22,29,31]and repeated every 2-3 years,although recommendations differ.Patients with JPS or PJS should be offered colectomy,either segmental or subtotal with IRA or restorative proctocolectomy with IPAA if the colorectal polyp burden is too high for endoscopic management or if cancer develops[21,22,29].A further indication for resection in this population may be severe bleeding from colonic neoplasia[21,22,29](Figure 2).
UPPER Gl ENDOSCOPY AND SURGERY
For some polyposis syndromes,upper GI surveillance is recommended due to a high risk of polyposis and/or cancer.In patients with a pathogenic variant inAPCassociated with FAP,esophagogastroduodenoscopy(EGD)is recommended from 25 years of age.It may be initiated earlier if colonic polyposis is present in the teenage years[20,24].In recent years,it has become more frequent to perform EGD with a cap-assisted forward viewing endoscope,which has been shown to be safe and visualize the papilla in most patients[32].
It is widely recommended to alter surveillance according to the Spigelman staging of polyps[33,34].Depending on the EGD findings and histopathological evaluation,total duodenectomy may be relevant in patients with FAP.As a guidance to the timing of duodenectomy,the Spigelman classification may used[20,24].Duodenectomy is recommended in patients with stage IV or evidence of cancer[20,24].
In some cases,it is recommended to perform a pancreas preserving total duodenectomy,which facilitates an easier endoscopic surveillance compared to post-Whipple procedure.It is proposed that EGD screening in patients with JPS should start in the early teenage years and be repeated every 2-3 years[20,22].Several upper GI resections may be relevant in patients with JPS,especially those who have a pathogenic variant inSMAD4since they have a higher risk of gastric polyposis and gastric cancer compared to JPS and pathogenic variants inBMPR1A[14].In the case of development of numerous and/or very large gastric polyps,partial or total gastrectomy is advised[21,22,29].
In patients with PJS,initial EGD is recommended at the same time as the initial colonoscopy,usually at age 8-10[20-22,29].The repeat interval should be based on endoscopic findings,but due to the increasing risk of polyposis with age,the interval should at minimum be every 2-3 years[20-22,29](Figure 2).Upper GI resections in patients with PJS should in general be segmental,although indications should be made with some restraint[21,22,29].It is recommended to perform the smallest possible,oncologically safe resection to prevent the risk of short bowel syndrome.Indications include suspicious lesions,repeated symptomatic bleeding and obstruction or intussusception caused by polyps[21,22,29].Small bowel resection in patients with JPS follow the same recommendations.It is advised to perform intraoperative enteroscopy when performing small bowel resections to evaluate the extent of polyposis.
OTHER ENDOSCOPlC PROCEDURES
In PJS,polyps mainly develop in the in the small bowel,and invagination is a frequent first symptom[21,22,29].It is usually recommended that patients with PJS undergo a baseline video capsule endoscopy at the beginning of endoscopic screening[21,22,29].Intervals for video capsule endoscopy should depend on the endoscopic findings.
CONCLUSlON
As more and more families are identified and genetic testing is becoming more sophisticated,research into preventing symptomatic disease has increased.In patients with FAP,the primary focus has been on nonsteroidal anti-inflammatory drugs,which have been shown to decrease the number of adenomas.Two agents in particular have been studied:Sulindac and celecoxib.Both have demonstrated a decrease in the amount of colorectal adenomas,but celecoxib also decreases the number of duodenal adenomas[28].Treatment with nonsteroidal anti-inflammatory drugs inherently has a risk of GI complications,most of which can be managed with protein-protein interaction.Celecoxib,a selective cyclooxygenase-2 inhibitor,has the disadvantage of an increased risk of cardiovascular side effects.Treatment with celecoxib has shown similar results in patients with PJS.Polyps in PJS overexpress cyclooxygenase-2,and the decrease in polyp burden is thought to be due to inhibition of this expression[20].In addition,some older studies have shown that rapamycin(sirolimus)affects the polyp burden and size in mouse models[35,36].Further therapeutics are under ongoing investigation and aim to target the involved pathway.In JPS,bothBMPR1AandSMAD4encode proteins working in the transforming growth factor-beta pathway,and as it is also frequently involved in sporadic cancer several attempts have been made to target this[37].Recommendations as to which patients should use chemoprevention differs,but since no agents have been shown to prevent development of disease,the endoscopic and surgical measures,as described above,should be the primary focus.
FOOTNOTES
Author contributions:Pachler FR and Jelsig AM drafted the manuscript;Pachler FR,Byrjalsen A,Karstensen JG and Jelsig AM provided revisions within their expert fields;Byrjalsen A drew up figures and tables.
Conflict-of-interest statement:Karstensen JG is a consultant for Snipr Biome.Pachler FR,Byrjalsen A and Jelsig AM have no conflicts of interest to declare.
Open-Access:This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers.It is distributed in accordance with the Creative Commons Attribution NonCommercial(CC BYNC 4.0)license,which permits others to distribute,remix,adapt,build upon this work non-commercially,and license their derivative works on different terms,provided the original work is properly cited and the use is noncommercial.See:https://creativecommons.org/Licenses/by-nc/4.0/
Country/Territory of origin:Denmark
ORClD number:Frederik R?nne Pachler 0000-0001-6941-9617;Anna Byrjalsen 0000-0002-3470-5995;John Gásdal Karstensen 0000-0001-9333-0399;Anne Marie Jelsig 0000-0002-0916-4517.
S-Editor:Wang JJ
L-Editor:Filipodia
P-Editor:Wang JJ
 World Journal of Gastrointestinal Surgery2023年1期
World Journal of Gastrointestinal Surgery2023年1期
- World Journal of Gastrointestinal Surgery的其它文章
- Application of ablative therapy for intrahepatic recurrent hepatocellular carcinoma following hepatectomy
- Postoperative adjuvant therapy for hepatocellular carcinoma with microvascular invasion
- Prognostic effect of excessive chemotherapy cycles for stage ll and lll gastric cancer patients after D2 + gastrectomy
- Development and validation of a novel nomogram for predicting overall survival in gastric cancer based on inflammatory markers
- New perspectives on robotic pancreaticoduodenectomy:An analysis of the National Cancer Database
- lmpact of body mass index in elderly patients treated with laparoscopic liver resection for hepatocellular carcinoma