
Fast-tracking regenerative medicine for traumatic brain injury
2020-01-02 01:17BrookeBonsackMattHeyckChaseKingsburyBlaiseCozeneNadiaSadanandanJeaYoungLeeCesarBorlongan
中國神經(jīng)再生研究(英文版) 2020年7期
Brooke Bonsack, Matt Heyck, Chase Kingsbury, Blaise Cozene, Nadia Sadanandan, Jea-Young Lee, Cesar V. Borlongan
Center of Excellence for Aging and Brain Repair, University of South Florida College of Medicine, Tampa, FL, USA
Abstract Traumatic brain injury remains a global health crisis that spans all demographics, yet there exist limited treatment options that may effectively curtail its lingering symptoms. Traumatic brain injury pathology entails a progression from primary injury to inflammation-mediated secondary cell death. Sequestering this inflammation as a means of ameliorating the greater symptomology of traumatic brain injury has emerged as an attractive treatment prospect. In this review, we recapitulate and evaluate the important developments relating to regulating traumatic brain injury-induced neuroinflammation, edema, and blood-brain barrier disintegration through pharmacotherapy and stem cell transplants. Although these studies of stand-alone treatments have yielded some positive results, more therapeutic outcomes have been documented from the promising area of combined drug and stem cell therapy. Harnessing the facilitatory properties of certain pharmaceuticals with the anti-inflammatory and regenerative effects of stem cell transplants creates a synergistic effect greater than the sum of its parts. The burgeoning evidence in favor of combined drug and stem cell therapies warrants more elaborate preclinical studies on this topic in order to pave the way for later clinical trials.
Key Words: clinical trials; combined therapy; inflammatory cascade; neuroinflammation; neuroprotection;neurotrauma; pharmacotherapy; preclinical studies; secondary cell death; stem cells
Inflammation Represents an Attractive Target for Treating Traumatic Brain Injury
Traumatic brain injury (TBI) refers to brain damage and dysfunction resulting from an external blow or jolt to the head, such as a puncture, blunt impact, or blast (Maas et al.,2008). Representing a considerable source of health and economic costs, TBI influences approximately 1.7 million lives per year (Acosta et al., 2015a). Additionally, the frequency of TBI-related military casualties has increased markedly due to escalated usage of explosives in modern warfare (Okie, 2005).In humans, TBI severity—categorized as mild, moderate, or severe—can be evaluated using medical imaging techniques and by assessing a patient's level of consciousness with the Glasgow Coma Scale (Lozano et al., 2015). Symptoms of mild TBI, including but not limited to headache, dizziness, fatigue,and nausea, may be temporary and clear up in the shortterm; however, severe TBI may lead to chronic neurological problems akin to the degenerative pathological hallmarks of Alzheimer's disease, Parkinson's disease, and dementia pugilistica (Colmano and Gross, 1971; Lozano et al., 2015). According to epidemiological data, suffering a TBI also elevates an individual's risk for developing Alzheimer's disease and Parkinson's disease later in life, but the mechanisms underlying these processes remain unclear (Lozano et al., 2015).
Although TBI was once exclusively deemed an acute injury, its pathology is now divided into the acute and chronic temporal phases. The relatively brief acute phase comprises the immediate neurostructural damage resulting from TBI,whereas the chronic phase is distinguished by rampant inflammation, secondary cell loss, and impaired neural function which may last for years and is finally irreversible(Werner and Engelhard, 2007; Acosta et al., 2015a). Harmful byproducts arising from the primary injury site, such as pro-inflammatory molecules and reactive species, become dispersed throughout the brain and cause cyclic cascades of neural cell damage (Zhao et al., 2005; Acosta et al., 2015a).The accumulation of such damage forms the penumbra—a region of dead and vulnerable brain tissue surrounding the primary injury core—which chiefly contributes to the risk for chronic evolution of cognitive symptoms (Zhao et al.,2005; Acosta et al., 2015a). However, due to the potentially broad window for effective treatment, alleviating this progressive penumbral cell loss represents a promising approach to improve long-term outcomes for TBI patients (Zhao et al.,2005; Acosta et al., 2015a).
The peri-impact area is a common pathological characteristic of chronic TBI which manifests as the tissue in the vicinity of the focal injury site displaying a myriad of intricate metabolic, immune, and cellular responses (Kumar and Loane, 2012; Lozano et al., 2015). In contrast, the specific pathological progression of these responses, such as their possible spread to remote brain regions, varies based on the location and severity of the TBI (Kumar and Loane, 2012;Lozano et al., 2015; Pabon et al., 2016). Intervention to mitigate the primary damage of a TBI — including necrosis, excitotoxicity, and the mangling of local neuronal, microglial,and vascular structure — is largely impractical, and use of safety equipment such as helmets and seatbelts constitutes the only feasible means of prevention (Hirschenfang et al.,1968; Kumar and Loane, 2012; Lozano et al., 2015). Thus,recent laboratory investigations have concentrated on the chronic symptoms of TBI in order to suppress secondary cell death mechanisms following the acute phase (Lozano et al.,2015). Due to the relatively delayed onset of inflammatory processes compared to other secondary cell death pathways within the injured brain, targeting post-TBI neuroinflammation has emerged as an especially attractive approach to reduce secondary cell loss and limit neurological deficits(Matsumoto et al., 1986; Offit et al., 1986; Lozano et al.,2015). While inflammation actually protects the injured brain during the acute phase of TBI, a prolonged inflammatory condition throughout the chronic phase significantly exacerbates neural cell death (Lozano et al., 2015). Indeed,an extensive body of evidence demonstrates that attenuating chronic inflammation confers neuroprotective effects (Lozano et al., 2015). Inflammation represents the net result of the competitive interaction between numerous pro-inflammatory (e.g., tumor necrosis factor alpha (TNF-α) and interleukin(IL)-1β) and anti-inflammatory (e.g., IL-10 and transforming growth factor beta (TGF-β)) molecules, and the shift from a protective to degenerative state following TBI further displays the complex pathology underlying neuroinflammation (Lozano et al., 2015).
After TBI, inflammation-mediated neuronal loss and edema are likely intensified due to disruption of the blood-brain barrier (BBB) (Shlosberg et al., 2010; Neuwelt et al., 2011).The BBB, a highly selective layer of endothelium surrounding cerebral vasculature, regulates the passage of bloodborne compounds into the brain, and the neuroprotective capacity of the BBB hinges upon its structural integrity and the health of its endothelial cells, both of which are maintained by astrocytes (Neuwelt et al., 2011). The physical trauma of TBI impairs BBB structure and function, heightening its permeability and permitting peripheral immune cells,along with exogenous proteins such as albumin, fibrinogen,and thrombin, to infiltrate the parenchymal tissue of the brain (Shlosberg et al., 2010; Neuwelt et al., 2011; Lozano et al., 2015). This foreign influx precipitates microglial activation, which, although initially functioning as a protective immune response, will continue without restriction and may become self-perpetuating unless BBB integrity is restored(Shlosberg et al., 2010; Neuwelt et al., 2011; Lozano et al.,2015). Moreover, pro-inflammatory substances such as cytokines, chemokines, prostaglandins, and free radicals are secreted by the intruding peripheral immune cells, amplifying the inflammatory cascade and further destabilizing the BBB (Lozano et al., 2015). Subsequently, large molecules and cells more readily penetrate the biological barricade, generating an osmotic gradient in the brain which leads to the formation of cerebral edema and the buildup of intracranial pressure (Neuwelt et al., 2011; Lozano et al., 2015). Furthermore, elevated levels of matrix metalloproteinase-9 (MMP-9), vascular endothelial growth factor (VEGF), and other molecules damage tight junctions, continuously inhibiting repair of the BBB (Guo et al., 1989; Lozano et al., 2015).
Within the injured brain, immunological processes become even more complex due to heterogeneous microglial activation (Lozano et al., 2015). More specifically, in response to their local microenvironment, activated microglia,which serve as the brain's resident macrophages under normal conditions, may differentiate into a pro-inflammatory M1 phenotype or an anti-inflammatory M2 phenotype which suppresses the activity of M1 microglia (Lozano et al.,2015), although it must be noted that this terminology has been disputed recently (Ransohoff, 2016). Therefore, in light of the diverse and complicated inflammatory pathophysiology of chronic TBI, techniques devised to downregulate pro-inflammatory mediators, upregulate anti-inflammatory mediators, and stimulate BBB recovery display significant therapeutic potential in laboratory and clinical settings(Lozano et al., 2015). Previously, we have evaluated the role of inflammation in TBI pathology and the potential of standalone pharmacotherapy and stem cell-based therapeutic options (Mashkouri et al., 2016). Here, we expand and update this review, culminating in advancing the potential application of combined drug and stem cell therapies.
This review was compiled using PubMed with sources within the last ten years, with an emphasis on the most recent, novel, and comprehensive papers. If the topic did not have relevant information within the last ten years, we used the most recent paper.
Pharmacotherapeutic Options for Reducing Inflammation
With the goal of reducing secondary neural cell death following TBI, a variety of anti-inflammatory drug therapies have been explored in recent laboratory investigations (Table 1). To this end, statins represent a class of drugs with a well-established safety record and discrete, manageable side effects. Traditionally considered a high cholesterol treatment,statins display anti-inflammatory and neuroprotective properties that may expand their applicability to TBI. A murine subarachnoid hemorrhage model demonstrates that pretreatment with rosuvastatin, a type of statin, may reverse the increases in proinflammatory TNF-α, MMP-9, and cyclooxygenase-2 usually observed after TBI (Uekawa et al., 2014).Adding to this, simvastatin reduces IL-1β inin vitrocultures of astrocytes (Wu et al., 2010). The mechanism behind the downregulation of these inflammatory mediators may be statins' inhibition of microgliosis and astrogliosis (Wu et al.,2010; Uekawa et al., 2014). Moreover, laboratory evidence of statins' regulation of epidermal growth factor receptors,some types of small G-proteins, and nuclear factor κB and toll-like receptor 4 signaling pathways may, in turn, suggest a mechanism behind these reductions in microgliosis and astrogliosis (Takemoto and Liao, 2001; Loane and Faden,2010; Wu et al., 2010; Uekawa et al., 2014; Wang et al., 2014).Thus, statins may sequester neuroinflammation by modulating several deleterious post-TBI processes. Furthermore,atorvastatin and simvastatin attenuate functional deficits and enhance neuronal survival in preclinical studies (Wang etal., 2007). Echoing this, rosuvastatin pretreatment protects against TBI-induced edema, neurological deficits, neuronal cell death, and BBB disruption in animal models, demonstrating that these anti-inflammatory effects may translate to improved system-wide outcomes (Uekawa et al., 2014).Such findings have helped rosuvastatin advance to a clinical trial, where multiple doses effectuated small reductions of disorientation and amnesia in TBI patients, as measured by the Galveston Orientation Amnesia Test (Tapia-Perez et al.,2008). More elaborate preclinical studies are necessary to assess how clinical administration and outcomes may be best optimized.
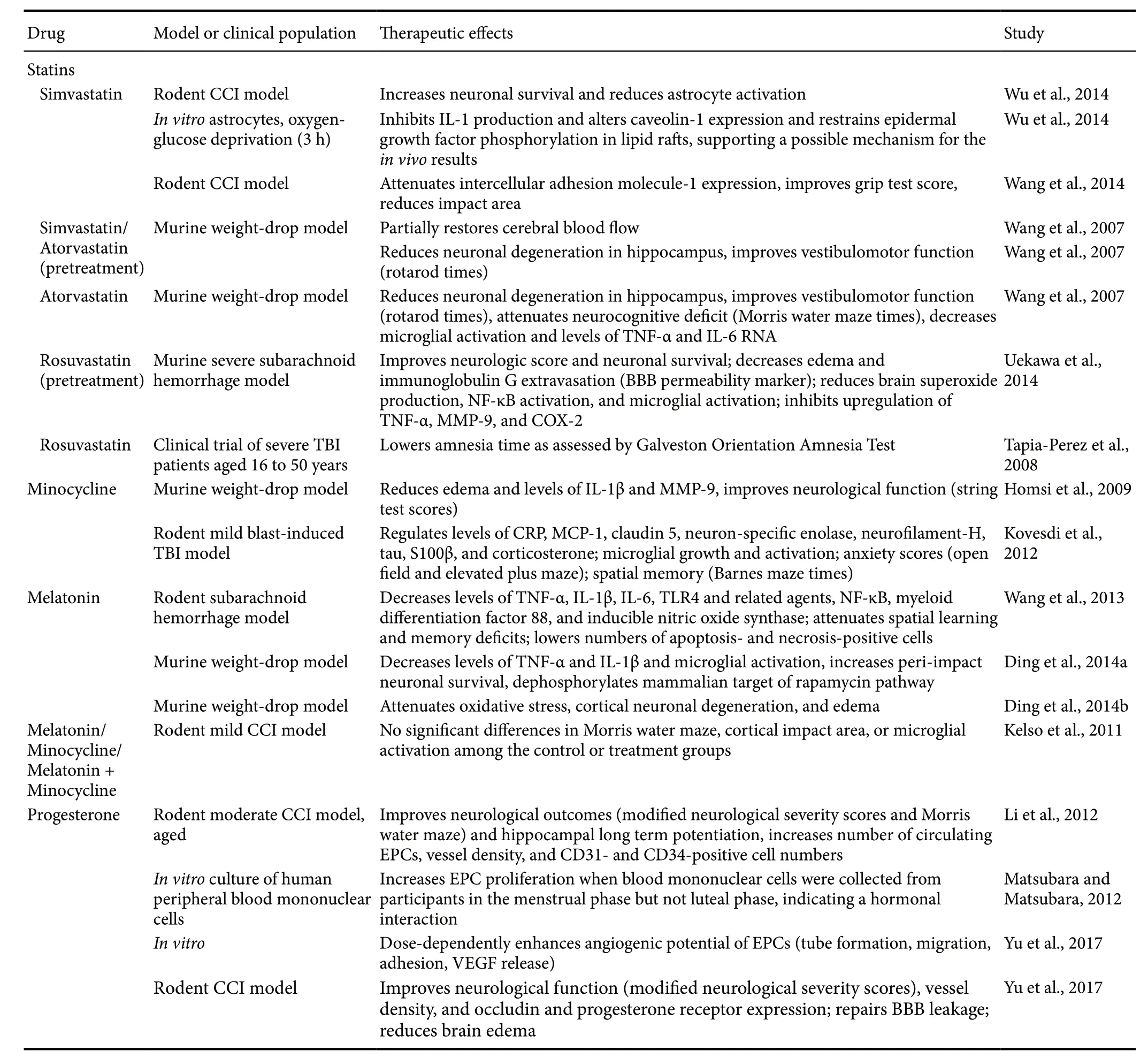
Table 1 Milestone studies of drug-based therapies for TBI
Aside from its antibiotic properties, minocycline, a tetracycline derivative, may also exert neuroprotective and anti-inflammatory effects. That it has a well-established record as a treatment for several diseases and is approved by the United States Food and Drug Administration advances its translation to TBI. Minocycline also possesses the ability to cross the BBB (Saivin and Houin, 1988), which promotes its use for central nervous system (CNS) disorders. These attributes posit minocycline as an excellent prospect for clinical trials. Expanding this background to TBI, minocycline treatment reduces edema and levels of inflammatory mediators IL-1β and MMP-9 in a murine weight-drop model (also known as closed head injury model) (Homsi et al., 2009)and normalizes levels of inflammatory markers C-reactive protein and monocyte chemoattractant protein-1 in a rodent model of mild blast-induced TBI (Kovesdi et al., 2012) when compared to vehicle treatment. Additionally, minocycline may lower nitric oxide, another inflammatory mediator, via its regulation of nitric oxide synthase, demonstrated in anin vitrostudy (Amin et al., 1996). These anti-inflammatory effects are coupled with a reduction in microglial growth and activation (Kovesdi et al., 2012). As microglia support cytokines MMP-9, IL-1β and IL-6, inhibiting microglial activation may indirectly sequester these cytokines as well(Homsi et al., 2009; Ziebell and Morganti-Kossmann, 2010;Guo et al., 2011). Furthermore, a string test, designed to assess neurological and locomotor functioning by investigating whether TBI mice can climb onto a string, walks, and escapes to a platform, indicates that these anti-inflammatory and anti-edematous effects can translate to functional recovery (Homsi et al., 2009). While the results of some laboratory studies have demonstrated null effects of minocycline on one or more of these dimensions, this may be due to inadequate dosage timing and amounts (Homsi et al., 2009; Kelso et al.,2011). As such, further investigation into minocycline's therapeutic potential is needed.
The neuroprotective and anti-inflammatory qualities of melatonin, a naturally-occurring hormone, similarly posit that this drug serves as another possible pharmacotherapy for TBI. Secreted by the pineal gland, melatonin possesses the ability to diffuse across cell membranes and cross the BBB (Lozano et al., 2015; Lin et al., 2016). Melatonin treatment is well-tolerated by patients and has a history of use for various other diseases (Jahnke et al., 1999; Seabra et al.,2000). Melatonin treatment after induced subarachnoid hemorrhage in rats reduces levels of proinflammatory cytokines TNF-α and IL-1β (Wang et al., 2013). Likewise, in a murine weight-drop model, the melatonin-treated group exhibits lower levels of these same proinflammatory cytokines when compared to the untreated and vehicle-treated groups (Ding et al., 2014b). These anti-inflammatory effects are coupled with melatonin's demonstrated ability to greatly inhibit microglial activation, as measured by IBA-1 immunofluorescence, a marker of microglia (Ding et al., 2014b).The same model also reveals that melatonin may ameliorate post-TBI neuronal degeneration, oxidative stress, and edema, indicating that melatonin could recapitulate cognitive function in humans (Ding et al., 2014a). However, similar to minocycline, these functional outcomes of melatonin are somewhat inconsistent. When administered alone or in concert with minocycline, melatonin had no effect on cortical tissue integrity or Morris water maze performance in one rodent model (Kelso et al., 2011). This may be due, however,to suboptimal timing and dosages. As it stands, melatonin has been implicated in multiple anti-inflammatory and neuroprotective processes, but these have, at times, failed to translate to functional benefits. To address this disparity,more studies examining the long-term safety and efficacy of melatonin as a TBI therapeutic are needed (Hirschenfang et al., 1968; Lozano et al., 2015).
Overall, pharmacotherapeutic options have demonstrated promise for attenuating TBI pathology, especially with respect to neuroinflammation. However, these results are at times somewhat tenuous or conflicting. Thus, other options,such as the rapidly emerging field of stem cell transplants,may be considered.
Stem Cell Therapy Ameliorates Inflammation
In addition to anti-inflammatory pharmacotherapy, stem cell therapy represents a promising approach for treating TBI(Table 2). Stem cells have the ability to proliferate and differentiate into a variety of cell lines and entice restorative cytokines to migrate to the injured area (Antonucci et al., 2014;Tajiri et al., 2014a, c). Preclinical data have revealed the effi-cacy of stem cells to differentiate into neurons after TBI and other CNS disorders (Antonucci et al., 2012; Rodrigues et al.,2012; Liu et al., 2013; Acosta et al., 2014; Tajiri et al., 2014a,b). Thus, direct cell-replacement constitutes one of the ways in which stem cells may attenuate TBI pathology. Secondly, stem cells may secrete various trophic factors that may directly or indirectly support beneficial processes while inhibiting deleterious ones, also known as “bystander effects”.Furthermore, evidence of a stem cell-paved “biobridge” by which exogenous stem cells may conduct endogenous stem cells to the impact area to decrease inflammation supports a third method by which stem cells may promote recovery after TBI (Tajiri et al., 2013). Interestingly, this biobridge links the injury site to the subventricular and subgranular zone,which primarily houses neural progenitor cells (Tajiri et al.,2013). This biobridge may thus encourage the migration and differentiation of adult neural progenitor cells. Overall, following brain trauma, stem cell transplantation exhibits a significant ability to ameliorate neural cell death and secondary inflammatory response, leading to improved cognitive and motor outcomes in preclinical settings (Acosta et al., 2014;De la Pena et al., 2014).
Stem cells' secretome plays a major role in their reduction of TBI-induced inflammatory response. Mesenchymal stem/stromal cells (MSCs) possess the ability to travel to the injury site and initiate the cellular effectors of the inflammatory response such as macrophages, neutrophils, T lymphocytes, and microglia (Borlongan, 2011; Borlongan et al., 2011; Zhang et al., 2013). Macrophages represent one component of the inflammatory response and function primarily through phagocytosis (Lin and Du, 2018). In a spinal cord injury model, embryonic stem cell-conditioned media facilitates macrophage phagocytosis of apoptotic cells, thus reducing inflammation and improving recovery (Guo et al.,2016). Moreover, when exposed to inflammatory factors,MSCs may secrete prostaglandin E2, which, in turn, may induce macrophages to release anti-inflammatory IL-10 and TGF-β (Németh et al., 2009; Chiossone et al., 2016), and which may also influence macrophages toward their anti-inflammatory M2 phenotype or initiate production of new M2 macrophages (Cho et al., 2014; Geng et al., 2014; Vasandan et al., 2016). In addition, promotion of neutrophils, cells that support healing and protect against infection, also holds potential for reducing inflammation. MSCs may secretechemokines that attract neutrophils, which in turn curtail inflammation (Brandau et al., 2010; Lin and Du, 2018). Upon recruitment, neutrophils exposed to MSC-derived IL-6 also survive for longer and display enhanced functioning (Lin and Du, 2018). Lastly, MSCs may release various factors to sequester and suppress T cells that would otherwise increase inflammation and secondary cell death (Lin and Du, 2018).Taken together, preclinical models evince that exogenous stem cells may enhance, inhibit, recruit, and generally adjust the activities of other cell types to reduce inflammation.
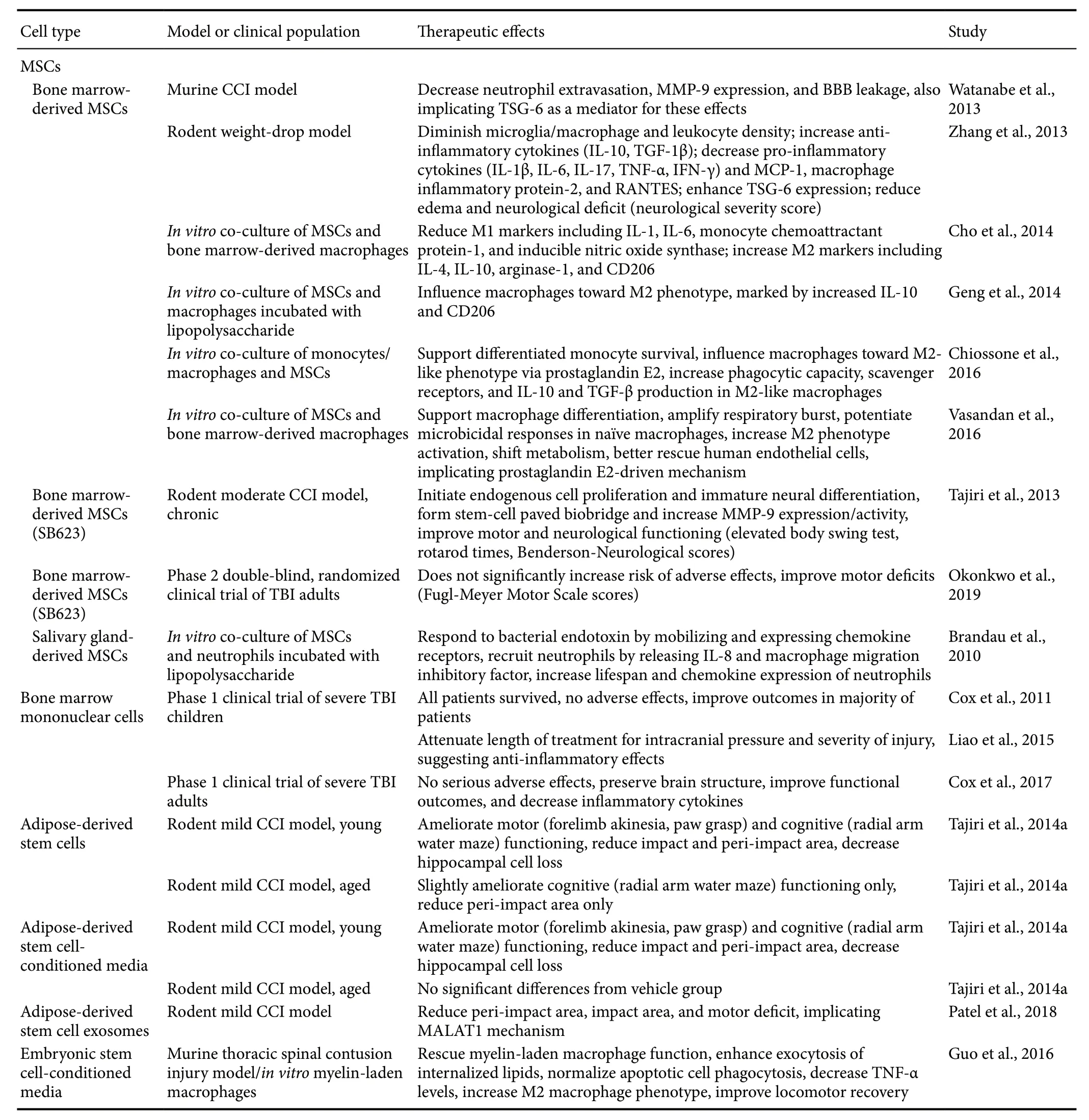
Table 2 Milestone studies of cell-based therapies for TBI
Aside from their regulation of other cells to indirectly modulate inflammation, stem cell transplants may also control the secondary cell death cascade by directly releasing anti-inflammatory agents. Key inflammatory signatures,such as TNF-α and IL-1, elicit the secretion of an anti-inflammatory protein known as TNF-α-stimulated gene/protein 6 (TSG-6) by the transplanted MSCs (Watanabe et al.,2013; Zhang et al., 2013). The anti-inflammatory property of TSG-6 acts by interrupting the inflammatory pathway of toll-like receptors and NF-κB (Watanabe et al., 2013; Zhang et al., 2013). Furthermore, TSG-6 can mitigate T cell activity by downregulating their production of pro-inflammatory cytokines, such as interferon γ and by upregulating anti-inflammatory cytokines, such as IL-4 (Russo et al., 2011).Thus, TSG-6 exemplifies one of the ways that stem cell therapy may abrogate neuroinflammation via their secretome.Another major way in which stem cells' secretome may reduce inflammation is through their emission of exosomes, a kind of nanoparticle that facilitates the release and transfer of proteins, DNA, mRNA and miRNA (Valadi et al., 2007;Altanerova et al., 2017). Long noncoding RNA metastasis-associated lung adenocarcinoma transcript 1 (MALAT1)is sometimes carried by exosomes originating from human adipose-derived stem cells and represents one of the beneficial agents that may be contained in exogenous stem cells'secretome (Patel et al., 2016, 2018). In brain and spleen tissues, treatment with exosomes containing MALAT1 regulates TBI-induced inflammatory pathways, regenerative pathways, cell cycle and death, and expression of other noncoding RNAs in a rodent controlled cortical impact (CCI)model (Patel et al., 2018). In turn, these MALAT1-controlled mechanisms generate significant post-TBI improvements in impact and peri-impact areas and on locomotor assessments,when compared to groups treated instead with MALAT1-depleted exosomes or with vehicle (Patel et al., 2018). Altogether, stem cell transplants possess multiple mechanisms whereby they may reduce inflammation through secreted factors.
Once the transplanted stem cells reach the host tissue,they are challenged by the harsh environment of the injury site. Granulocyte-colony stimulating factor (G-CSF) can be administered alongside human umbilical cord blood cells(hUCBs) to promote the neuroprotection of the exogenous stem cells (Acosta et al., 2014). Delivering stem cells in combination with factors such as G-CSF yields significant improvements in neurogenesis and the capacity to reduce cell death, when compared to delivering stem cells alone(Acosta et al., 2014). Moreover, G-CSF possesses properties that reduce brain edema, promote the recovery of motor function, and enhance control of glutamate levels (Acosta et al., 2014). G-CSF uses a receptor-mediated transport to mobilize endogenous stem cells from the bone marrow into the peripheral blood. Once in the periphery, the cells can then relocate to the injury site and promote the release of growth factors, chemokines, and cytokines that assist in repairing brain tissue (Acosta et al., 2014). Recent preclinical evidence that stem cells migrate to and survive better in the spleen rather than the brain suggests that direct transplantation may not be as efficacious as a peripheral approach (Acosta et al., 2015b). In fact, there are studies emphasizing systemic delivery of stem cells in TBI and other neurodegenerative models (Acosta et al., 2015b).
Several transplantable cell lines have been tested, with some reaching clinical trials for stroke therapy, such as fetal cells, embryonic stem cells, neural stem/progenitor cells(including NT2N and CTX0E3 cells), umbilical cord blood cells, amnion cells, adipose cells, and induced pluripotent stem cells (Hara et al., 2008; Li et al., 2008; Stroemer et al.,2009; Kaneko et al., 2011; Liu et al., 2014; De la Pena et al.,2015). Due to its robust safety record in other disease models, bone marrow and its cellular derivatives have been given a particular interest (Borlongan et al., 2011; Steinberg et al.,2016). Various preclinical studies also support that bone marrow-derived stem cells are not only safe, but also effective, in that bone marrow-derived mononuclear cells may preserve the BBB, promote neurogenesis, and facilitate functional outcomes (Cox et al., 2017). Furthermore, different routes of transplantation such as intravenous, intra-arterial,and intranasal as well as direct intracerebral implantation have all demonstrated the functional benefits bone marrow-derived stem cells offer (Borlongan et al., 2004, 2011;Prasad et al., 2014; Acosta et al., 2015b). Such an auspicious record has given researchers the impetus to explore bone marrow mononuclear cells and bone marrow-derived stem cells in clinical studies, where they have demonstrated no difference in adverse events, though further investigations are warranted to evaluate their efficacy (Cox et al., 2011,2017; Liao et al., 2015; Okonkwo et al., 2019). This notion calls attention to the need for combination therapy in order to improve the outcomes of pharmacotherapeutics and stem cell transplantations in TBI and other neurodegenerative diseases.
Current Efforts in Combined Drug and Stem Cell Therapies for Traumatic Brain Injury
Thus far, the reviewed studies have focused on the status of either stand-alone stem cell transplants or certain pharmacotherapeutics. Interestingly, combinations of the two are emerging as the most promising TBI therapeutics (Figure 1).A systematic review of combination therapies for TBI demonstrates the wide variety of potential treatments and groups the results by whether the combination yields enhanced,unaffected, or diminished effects as compared to stand-alone therapies (Kline et al., 2016). Therapies combining stem cell transplants with drugs and other supplements have produced auspicious results, possibly due to the amenability of stem cells to combined use and the ability of some drugs and supplements to facilitate and optimize the ameliorative actions of stem cells (Table 3).
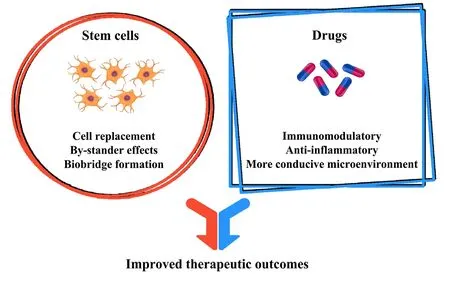
Figure 1 Drugs and stem cells work together to yield enhanced therapeutic outcomes.During the chronic phase of traumatic brain injury, persisting neuroinflammation contributes significantly to secondary neural cell death. While stand-alone cell therapy can be effective to an extent, combining stem cell transplantation with drug therapies may synergistically attenuate inflammatory neurodegeneration and promote regenerative mechanisms within the brain, thereby enhancing functional recovery following traumatic brain injury.
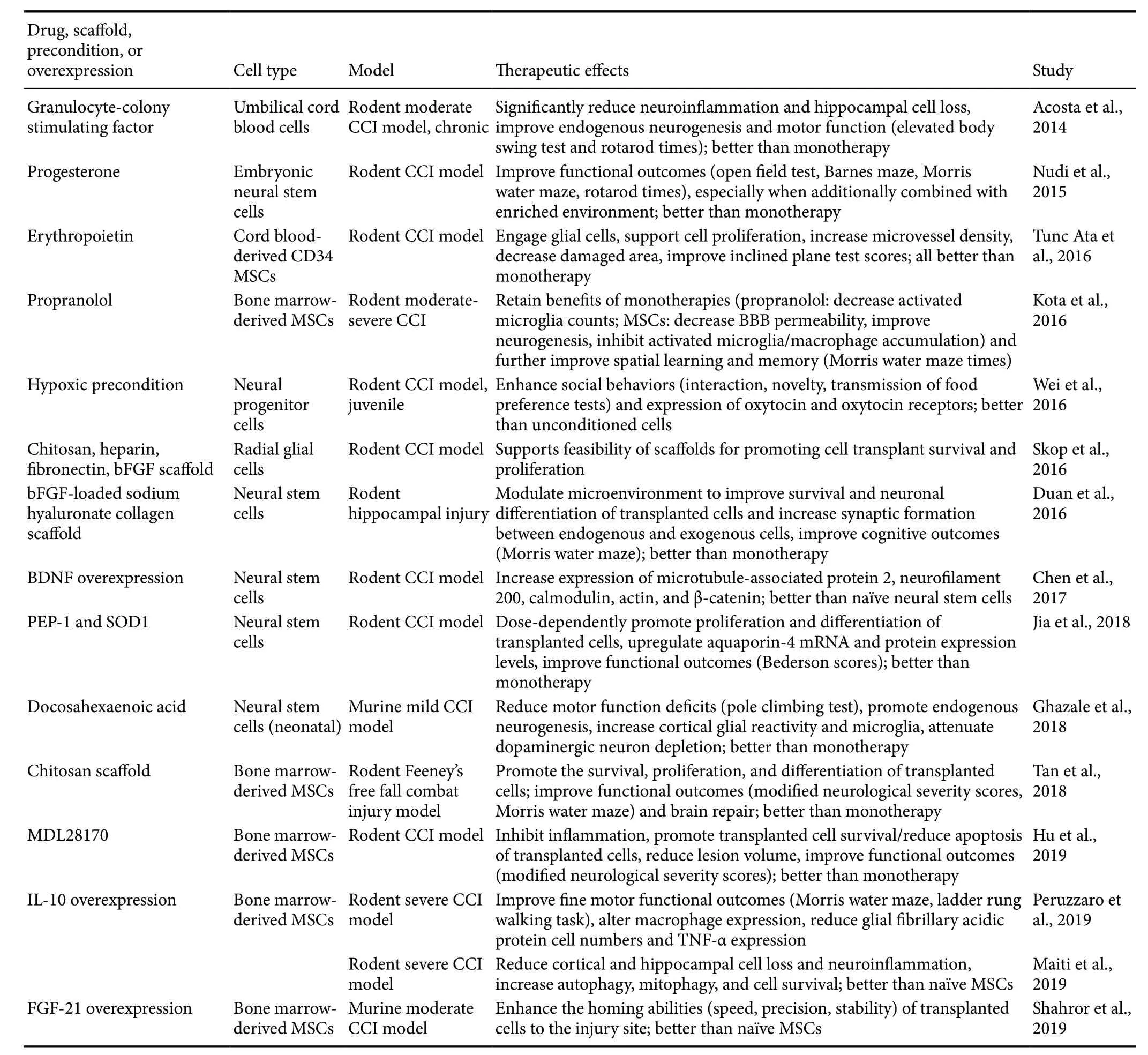
Table 3 Milestone studies of combination therapies for TBI
Many hormones may produce benefits when administered alone or in concert with stem cells. The sex hormone progesterone has many applications for various CNS disorders due to its penchant for anti-inflammatory and neuroprotective effects. Moreover, that it may encourage the proliferation of endothelial progenitor cells (EPCs) and improve post-TBI neuroregeneration advances progesterone as an attractive option for combination with stem cells (Li et al., 2012; Matsubara and Matsubara, 2012). In anin vitromodel, administering progesterone to rat EPCs dose-dependently increases secretion of angiogenesis precursors, including VEGF (Yu et al., 2017). Furthermore, as demonstrated by a CCI model of TBI, male rats who receive intraperitoneal progesterone exhibit enhanced vessel density, occludin expression, and BBB integrity, as well as reduced edema and neurological deficit scores, as compared to vehicle-treated rats (Yu et al.,2017). Additionally, that a progesterone receptor-antagonist reverses all of these benefits indicates that progesterone is a key facilitator of endogenous EPC brain repair (Yu et al.,2017). To further support that it is the interaction between progesterone and stem cells that produces these benefits,another study using the CCI model was performed with a double control. Combined therapy of embryonic neural stem cell (eNSC) transplants and progesterone begets improved performances on Morris water maze, open field, and rotarod functional assessments when compared to the control treatment, progesterone-only treatment, or eNSC-only treatment,as well as enhanced survival, migration, and differentiation by the stem cells when compared to the eNSC-only treatment (Nudi et al., 2015). Taken together, these studies indicate that progesterone may greatly stimulate and enable the neuroprotective and anti-inflammatory effects of stem cells,although more double-controlled studies using alternate TBI models are needed (Zibara et al., 2019).
Another naturally-occurring hormone, erythropoietin(EPO), helps to produce red blood cells under normal conditions but may also be applied to TBI treatment because of its neurotrophic, angiogenic, anti-inflammatory, and anti-apoptotic effects (Gonzalez et al., 2007; Bath et al., 2013).In a rodent model, combining EPO treatment with cord blood-derived CD34 MSCs yields enhanced post-TBI cell proliferation, neuronal damage attenuation, glial cell activation, microvessel density, impact area healing, and functional recovery beyond that which is produced by EPO-only or stem cell-only treatment (Tunc Ata et al., 2016). Thus, this demonstrates that EPO and stem cell combination therapy is another promising area for further research.
Various other pharmacotherapeutics may be combined with stem cells to attenuate TBI pathology. Some of these drugs, including G-CSF; peptide carrier PEP-1; antioxidant enzyme Cu,Zn-superoxide dismutase (SOD1); and polyunsaturated fatty acid docosahexaenoic acid (DHA), primarily work by synergizing with stem cells to directly stimulate and support beneficial endogenous processes. As previously discussed, co-administration of G-CSF with hUCBs may confer neuroprotective and anti-inflammatory effects greater than those conferred by stand-alone G-CSF or hUCBs in a CCI model of moderate TBI, possibly due to G-CSF's recruitment of stem cells to the injury site and its suppression of MHCII microglia activation (Acosta et al., 2014; Liska and de la Pena, 2017). Furthermore,in vitromeasures of PEP-1 and SOD1's effects on neural stem cells (NSCs) reveal that these drugs may dose-dependently increase NSC proliferation in a rodent model (Jia et al., 2018). This is supported by further tests demonstrating that co-administering PEP-1, SOD1 and NSCsin vivoproduces higher levels of brain aquaporin-4,which is beneficial for regulating edema in chronic TBI, and improves neurological function, as quantified by the modified Benderson score, when compared to vehicle treatments and stand-alone treatments (Jia et al., 2018). In addition,DHA may exhibit neuroprotective properties when combined with NSC transplants. Co-administering these two in a CCI murine model increases neurogenesis, glial reactivity,and dopaminergic neurons; decreases calpain/caspase activation; and improves motor function scores, as measured by the pole climbing test, when compared to vehicle treatments and NSC-only treatments (Ghazale et al., 2018).
Other drugs, including propranolol, a β-adrenergic receptor antagonist, and MDL28170, a calpain inhibitor, primarily work by suppressing deleterious factors that may interfere with endogenous or stem cell transplant-mediated reparative processes. Combining propranolol with later administration of human bone marrow-derived MSCs (BM-MSCs) yields complementary benefits greater than either treatment by itself in laboratory settings (Kota et al., 2016; Dekmak et al., 2018). Along with other improvements commensurate with the stand-alone therapies, combined treatment produces greater recovery of cognitive and memory faculties, as compared to vehicle treatments and stand-alone treatments(Kota et al., 2016). Furthermore, MDL28170 facilitates BMMSC survival and reduces BM-MSC apoptosis after transplantation in a rodent model (Hu et al., 2019). Thus, after experimental TBI, administering MDL28170 followed by BM-MSCs renders lower levels of inflammation, reduces lesion volume, and improves neurological function scores as compared to the vehicle treatment and the stand-alone treatments (Hu et al., 2019). Taken together, many drugs may confer complementary and additive effects when combined with stem cell transplants by enhancing positive processes or by suppressing negative processes.
While the synergy of stem cells and anti-inflammatory drug therapies may represent the most direct approach to counteracting secondary neurodegeneration in chronic TBI,the primary hindrance to stem cell transplants continues to be ensuring their adequate migration, survival, proliferation,and differentiation within the hazardous environment of the TBI brain (Skop et al., 2016; Zibara et al., 2018). Therefore, a number of studies have employed the use of biocompatible scaffolding to more effectively deliver and secure stem cells at injury sites. For example, radial glial cells fixed within an extensively interlinked framework of chitosan, fibronectin,heparin, and basic fibroblast growth factor may be implanted and successfully incorporated into the brains of TBI rats(Skop et al., 2016). Similarly, BM-MSCs complexed with chitosan porous scaffolds can be directly delivered into the lesions of TBI rats without adverse effects (Tan et al., 2018).In fact, this combination treatment increases the survival,proliferation, and differentiation of the BM-MSCs, thereby improving functional recovery (Tan et al., 2018). Moreover,joint transplantation of NSCs and a basic fibroblast growth factor-loaded sodium hyaluronate collagen scaffold enhances the survival of NSCs as well as their differentiation into functional neurons within injured regions of the TBI rat brain (Duan et al., 2016). Along with these effects, the promotion of synaptogenesis in the presence of this scaffold further contributes to the significant amelioration of cognitive deficits (Duan et al., 2016).
In addition to combinatorial treatments with cell-based therapy and pharmaceuticals or scaffolding biomaterials,other recent preclinical studies reveal that modifying the behavior and secretome of stem cell transplants may significantly enhance their anti-inflammatory effects, as well as their migration, viability, and potency in injured regions of the brain. In the context of reprogramming the behavioral aspect of stem cells for TBI treatment, preconditioning NSCs in a hypoxic environmentin vivobefore transplantation activates cell survival mechanisms which prime their resistance to the hostile conditions of the damaged brain (Wei et al., 2016; Zibara et al., 2018). Compared to NSCs cultured in healthy conditions prior to infusionin vivo, hypoxia-preconditioned NSCs more effectively attenuate inflammation,elevate release of trophic factors (i.e., glial cell line-derived neurotrophic factor, VEGF, brain-derived neurotrophic factor (BDNF), and EPO), and promote neurological recovery in a juvenile rodent model (Wei et al., 2016). In specific regard to augmenting the secretory functions of exogenous stem cells, inducing the overexpression of various trophic factors has proven to be promising therapeutic approach.Notably, genetically engineered BM-MSCs overexpressing the anti-inflammatory cytokine IL-10 increase the prevalence of autophagy, mitophagy, and cell-survival markers in a rat model, indicating the protection of neural cells from inflammation and oxidative stress (Maiti et al., 2019). These modified BM-MSCs also exert immunomodulatory effects,likely by altering inflammatory cell marker expression by macrophages, which may produce an environment that is more conducive to brain repair and functional recovery after experimental TBI (Peruzzaro et al., 2019). Indeed, MSCs induced to overexpress FGF-21 with this technique display more accurate migratory homing to TBI lesions in a murine model (Shahror et al., 2019). Furthermore, NSC transplants excessively producing BDNF exhibit improvements in survival, growth, proliferation, and differentiation into neurons at lesion sites in a rodent CCI model, potentially due to BDNF-induced upregulation of cytoskeletal protein expression(Chen et al., 2017; Zibara et al., 2018).
Overall, stem cell-based combination therapies stand as attractive strategies for TBI treatment. As previously mentioned, one review categorizes a wide variety of TBI-targeted combination therapies and classifies them by whether the combination produces enhanced, null, or reduced effects(Kline et al., 2016). Among the categories, studies of stem cell-based combination therapies consistently generate the highest proportion of enhanced effects and the lowest proportion of diminished effects when compared to studies of combination therapies not involving stem cells (Kline et al., 2016). Nevertheless, recent research of stem cell-based combination therapy for TBI is somewhat limited. This lack of studies is difficult to explain in light of stem cell-based combination therapies' relative success. The findings of stem cell-based combination therapies are promising, and thus warrant greater examination to advance these therapies to clinical trials.
Conclusion
In this review, a plethora of studies outline different treatment regimens for TBI with pharmacotherapy, cell therapy,or a combination of the two. Throughout the chronic phase of TBI, cyclically perpetuated neuroinflammatory cascades—characterized by upregulated levels of pro-inflammatory molecules, downregulated levels of anti-inflammatory molecules,and dysfunction of the BBB—cause devastating secondary cell death in the brain. While other mechanisms contribute to this neurodegeneration, the delayed onset and long-term progression of inflammatory processes are indicative of a wide therapeutic window for intervention. Thus, reducing aberrant neuroinflammation represents a promising approach to ameliorate secondary cell loss and promote neurological and functional recovery in TBI victims. Several pharmacological therapies demonstrate anti-inflammatory effects; however,inadequate dosages and poor clinical models have hindered their observed efficacy as TBI treatments. Instead, stem cells have attracted the attention of clinicians due to their capacity to replace lost brain cells, secrete neurotrophic factors, and recruit restorative cytokines and endogenous stem cells to the injured area. Furthermore, a combination of both pharmacotherapeutics and stem cells has yielded the most favorable outcomes. Certain drugs amplify the neuroregenerative properties of stem cells and provide a more suitable environment for the cells of the brain. Altogether, further investigation is needed to optimize treatment parameters and ensure safety in larger animal models, as well as to design more effective controlled small animal studies, before stem cell combination therapy can be translated to the clinic.
Author contributions:Drafting, editing, and formatting the manuscript:BB, MH, CK, and CVB; figure creation: CK and JYL; literature retrieval and collection: BC and NS. All authors approved the final manuscript.
Conflicts of interest:CVB was funded and received royalties and stock options from Astellas, Asterias, Sanbio, Athersys, KMPHC, and International Stem Cell Corporation; and also received consultant compensation for Chiesi Farmaceutici. He also holds patents and patent applications related to stem cell biology and therapy. The other authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Financial support:CVB was funded by National Institutes of Health(NIH) R01NS071956, NIH R01NS090962, NIH R21NS089851, NIH R21NS094087, and Veterans Affairs Merit Review I01 BX001407.
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-Non-Commercial-ShareAlike 4.0 License, which allows others to remix, tweak,and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewer:Elizabeth D. Kirby, The Ohio State University,USA.
Additional file:Open peer review report 1.
- 中國神經(jīng)再生研究(英文版)的其它文章
- Glial cells in intracerebral transplantation for Parkinson's disease
- The N-formyl peptide receptors: contemporary roles in neuronal function and dysfunction
- Adrenomedullin: an important participant in neurological diseases
- Shifting equilibriums in Alzheimer's disease: the complex roles of microglia in neuroinflammation,neuronal survival and neurogenesis
- ABC efflux transporters at blood-central nervous system barriers and their implications for treating spinal cord disorders
- Biomaterials and neural regeneration