
Adrenomedullin: an important participant in neurological diseases
2020-01-02 01:17FengJiaoLiSiRuZhengDongMeiWang
中國神經(jīng)再生研究(英文版) 2020年7期
Feng-Jiao Li, Si-Ru Zheng, Dong-Mei Wang
College of Life Sciences, Laboratory of Neuroendocrinology, Provincial Key Laboratory of Developmental Biology and Neuroscience, Fujian Normal University, Fuzhou, Fujian Province, China
Abstract Adrenomedullin, a peptide with multiple physiological functions in nervous system injury and disease,has aroused the interest of researchers. This review summarizes the role of adrenomedullin in neuropathological disorders, including pathological pain, brain injury and nerve regeneration, and their treatment.As a newly characterized pronociceptive mediator, adrenomedullin has been shown to act as an upstream factor in the transmission of noxious information for various types of pathological pain including acute and chronic inflammatory pain, cancer pain, neuropathic pain induced by spinal nerve injury and diabetic neuropathy. Initiation of glia-neuron signaling networks in the peripheral and central nervous system by adrenomedullin is involved in the formation and maintenance of morphine tolerance. Adrenomedullin has been shown to exert a facilitated or neuroprotective effect against brain injury including hemorrhagic or ischemic stroke and traumatic brain injury. Additionally, adrenomedullin can serve as a regulator to promote nerve regeneration in pathological conditions. Therefore, adrenomedullin is an important participant in nervous system diseases.
Key Words: adrenomedullin; brain injury; glia; mechanism; morphine tolerance; neural regeneration;neuroprotective effect; pathological pain; regeneration; sensitization; target
Introduction
Neurological disorders such as trigeminal neuralgia (Chang et al., 2019; Chen et al., 2019; Sung et al., 2019), pathological pain (Campos et al., 2019), multiple sclerosis (Schirmer et al., 2019), senile dementia (Zulfiqar et al., 2019), stroke(Huang et al., 2019b) and neurotic tinnitus (Mulders et al.,2019) affect a large number of patients worldwide. Pathological pain, also known as chronic pain, is a common clinical neurological disease that includes inflammatory, neuropathic and cancer pain (Odell, 2018). Paralgesia is a manifestation of pathological pain and includes hyperalgesia, allodynia and spontaneous pain. Inflammatory pain refers to the pain that occurs when inflammation is caused by traumatic bacterial or viral infections, or peripheral tissue damage caused by surgery. Neuropathic pain is induced by nerve damage, viral infections such as herpes zoster, or metabolic disorders such as diabetic neuropathy (Batista et al., 2019). In most cases,neuropathic pain will not be alleviated with remission of the primary disease (Zhou et al., 2018). Cancer pain is caused by compression or infiltration by tumors. Under various pathological pain conditions, because of the interaction between neurons and the immune system, the sensory nervous system suffers from maladaptive plasticity (Baskozos et al.,2019). Inflammation or peripheral nerve injury induces local release of inflammatory substances, stimulates peripheral nerve endings (Pogatzki-Zahn et al., 2019), induces upregulation of pain-related mediators or receptors in dorsal root ganglia (DRG) (Li et al., 2018), increases cell excitability and subsequently sensitizes cells (peripheral sensitization). This in turn increases the release of neurotransmitters at the central end of afferent fibers of DRG neurons (Petho et al., 2017)and increases the excitability of and ultimately sensitizes spinal dorsal horn neurons (central sensitization). Peripheral sensitization and central sensitization increase pain sensitivity and produce chronic pain. Opioids are the most effective analgesic treatments for this condition at present. However,long-term use of opioids can cause opioid tolerance and many side effects (Bobeck et al., 2019), such as nausea, vomiting and constipation (Daoust et al., 2019). Some known mechanisms of opioid tolerance are similar to those of pathological pain. Further study on the mechanism of morphine tolerance will undoubtedly be beneficial for the treatment of pathological pain. Brain injury, including traumatic and acquired brain injury (Song et al., 2019), is another neurological disorder with an increasing number of patients worldwide and is also a medical problem because its mechanism is unclear (Rucker et al., 2019). Nerve regeneration is an important treatment for many neurological diseases. This review focuses on the evidence implicating adrenomedullin (AM) in functional impairment of the nervous system caused by nervous system injury or neuropathic conditions,including pathological pain and brain injury. This review examines the progress in the use of AM for nerve regeneration and discusses the possible roles of AM in neurological diseases and its mechanism as a target for treatment.
Search Strategy
The articles included in this review were retrieved by an electronic search of the Medline database for literature describing the role of AM in neuropathological disorders using the following strategy: Adrenomedullin (MeSH Terms)AND Pain (MeSH Terms) OR Brain injury (MeSH Terms)OR Regeneration (MeSH Terms). Articles on AM and pain:the results were further screened by title and abstract to only present articles on pain types (inflammatory pain, neuropathic pain or cancer pain) and pain conduction-related sites(spinal and dorsal root ganglion and brain). Articles on AM and regeneration: the results were further screened by title and abstract to only present articles on the peripheral and central nervous system. Non-nervous system articles were excluded. In addition, an electronic search of the Medline database was performed using the following strategy: Adrenomedullin (MeSH Terms) AND Morphine tolerance (MeSH Terms). Furthermore, the following search strategy was performed: Adrenomedullin (MeSH Terms) and Multiple sclerosis (MeSH Terms) OR Huntington (MeSH Terms) OR Parkinson's disease (MeSH Terms) OR Alzheimer's disease(MeSH Terms).
Adrenomedullin
AM belongs to the calcitonin gene related peptide (CGRP)family, which includes CGRP, calcitonin, intermedin (IMD/AM2), amylin and calcitonin receptor-stimulating peptide(Baskaya et al., 1995). AM was discovered in 1993 in human pheochromocytoma (Kitamura et al., 1993). It is a 52-amino acid peptide containing a disulfide bond and is derived from a preprohormone (Kitamura et al., 1993). The human AM gene is located on chromosome 11 (Ishimitsu et al., 2001)and is expressed in many tissues, including the heart, eye,adrenal gland, kidney, liver, lung, thyroid and brain (Chen et al., 1997). AM exerts its bioactivity by binding to a receptor that consists of a heterodimer composed of the calcitonin receptor-like receptor and receptor activity-modifying protein type 2 (RAMP2) or 3 (RAMP3), which are called AM1 and AM2 receptors, respectively (Husmann et al., 2003). AM mainly acts on AM1 receptor (Hay et al., 2004). Initially, AM was identified as a vasoactive peptide, but it has also been shown to play a role in various physiological and pathological activities, including bronchodilatation, diuresis promotion, food intake, hormone regulation, gastrointestinal modulation, cell apoptosis, immune regulation, tumor progression and diabetes (Mak et al., 2006; Andren-Sandberg,2016; Arrigo et al., 2019; Bech et al., 2019). The involvement of AM in neurological diseases is gradually being explored,and AM has been shown to function as an inflammatory agent, a neuromodulator, a neurotransmitter and a neurohormone through autocrine or paracrine mechanisms. AM and its receptors are widely distributed throughout and play an extensive role in the nervous system.
Adrenomedullin and Pathological Pain
Involvement of adrenomedullin in inflammatory,neuropathic and cancer pain
In recent years, the important contribution of AM to pain has attracted much attention from researchers. AM is a powerful pain-inducing neuropeptide. AM mRNA and protein are abundantly localized in small-to-medium DRG neurons and laminae I-II of the spinal cord. Approximately 33% of AM-expressing DRG neurons are CGRP-positive, and up to 56% of IB4-binding neurons exhibit AM immunoreactivity(Ma et al., 2006). AM1 receptors are also distributed in the DRG and superficial spinal dorsal horn. AM coexists with its receptor components, calcitonin receptor-like receptor,RAMP2 and RAMP3, in small and medium DRG neurons(Hong et al., 2010). These findings suggest that AM may play a role at the DRG and superficial spinal dorsal horn, which are two important pain information transmission sites.Indeed, intrathecal injection of the AM1 receptor agonist AM1-50 produces thermal hyperalgesia (Ma et al., 2006)and mechanical allodynia (Huang et al., 2019a), which can be inhibited by the selective AM antagonist AM22-52. A complex relationship exists between AM and pain sensitivity.Knockout of the AM gene in central nervous system neurons will prolong the latency in the tail-flick test and shorten the latency in the hotplate test. The tail-flick test is an assessment of nociception, which is mediated predominantly by a spinal mechanism, and the hotplate test is a measure of nociception, which is mediated by supraspinal mechanisms.Therefore, AM acts as a nociceptive peptide for spinal reflexes but has analgesic properties when the brain is involved in pain processing (Fernandez et al., 2010). One study found that AM is not expressed by nonpeptidergic nociceptors in mice (Fernandez et al., 2010), illustrating a clear difference that seems to exist in the distribution of AM among species.Moreover, this effect of AM is achieved by upregulating CGRP in the DRG and spinal cord and downregulating substance P and enkephalin in the DRG, resulting in modulation of learning, adaptation and habituation. This finding suggests that lack of AM leads to changes in pain sensitivity. However, another study found that the AM antagonist AM22-52 did not produce this effect (Ma et al., 2006; Huang et al., 2019a). Therefore, whether AM participates in physiological pain is still controversial.
AM participates in pathological pain including acute and chronic inflammatory pain. Intrathecal administration of AM22-52 inhibits the hyperalgesia caused by capsaicin- (Ma et al., 2006), formalin- (Sugimoto et al., 2013) or complete Freund's adjuvant-induced inflammation (Hong et al., 2009).Meanwhile, acute and chronic inflammation can increase AM expression in spinal dorsal horn and small- and medium-sized neurons in the DRG (Hong et al., 2009; Sugimoto et al., 2013).
The inhibitory effect of AM22-52 on complete Freund's adjuvant-induced hyperalgesia lasts for at least 24 hours(Wang et al., 2013a). Similarly, AM1-50 also induces longterm hyperalgesia lasting longer than 24 hours (Ma et al.,2006). This is different from the transient pain-inducing properties of other substances such as CGRP (Hirsch et al.,2013) and may involve the ability of AM to recruit other pronociceptive mediators. Blocking of AM receptors inhibits the increase in CGRP and neuronal nitric oxide synthase (nNOS)induced by chronic inflammation (Wang et al., 2013a). Exogenous application of AMin vitrocan induce production of CGRP (Hong et al., 2010) and nNOS in the DRG (Wang et al., 2013a). AM does not induce production of nNOS in the DRG after inhibition of protein kinase A (PKA) activity. The effect of AM on inducing CGRP production in the DRG also disappears after inhibition of nNOS. This implies that complete Freund's adjuvant induces an AM-nitric oxide(NO)-CGRP cascade via the cyclic adenosine monophosphate (cAMP)/PKA signaling pathway (Wang et al., 2013a).Cancer pain is an intractable problem in clinical practice because even high doses of the strongest painkillers do not alleviate the pain. One study indicated that enhanced biological activity of AM is involved in the pathogenesis of bone cancer pain (Hu et al., 2018). Blockade of AM receptors by intrathecal AM22-52 reversed bone cancer-induced mechanical allodynia. Bone cancer causes an increase in AM protein and mRNA expression in the spinal cord and DRG,and these changes could be abrogated by intrathecal administration of AM22-52. Blocking of the AM receptor also inhibits expression of NOS and CGRP in the spinal cord and DRG (Hu et al., 2018), and chemokine (C-C motif) ligand 2(CCL2) is increased in the DRG (Chen et al., 2017) because of bone cancer. These findings indicate that AM participates in inflammatory and cancer pain and, as an important nociceptive modulator, can recruit other inflammatory regulators during pathological conditions. Moreover, AM is a relatively upstream inflammatory mediator and may trigger a sequence of events. It is also important for the early events during inflammatory pain. Early disruption of this cascade or cycle will undoubtedly provide a better means for earlier and more effective treatment interventions.
The substantial role of AM in inflammatory and cancer pain has led researchers to continue to explore the relationship between AM and other types of pathological pain. One laboratory group (unpublished data) found that AM is also involved in neuropathic pain induced by spinal nerve ligation. Spinal nerve ligation induced upregulation of AM in the peripheral (nerves and the DRG) and central nervous system;blockade of AM reduced the development and maintenance of neuropathic pain induced by spinal nerve ligation, and the mechanisms of AM in early and late neuropathic pain induced by these spinal nerve ligation were diverse. In the central nervous system, AM initiates neuropathic pain by activating microglia, while neuropathic pain is maintained by activating astrocytes. Peripheral AM participates in spinal nerve ligation-induced neuropathic pain by inhibiting NOS and transient receptor potential cation channel subfamily V member 1 (TRPV1) in the DRG. This group also found that AM plays a significant role in lipopolysaccharide-induced inflammatory pain and diabetic neuropathy. P2X7-dependent activation of satellite glia is a key mechanism by which AM facilitates both types of pain (unpublished data). As a powerful pain mediator, AM plays a key role in central and peripheral sensitization caused by pathological pain (Figure 1).
Involvement of adrenomedullin in morphine tolerance
Opioid tolerance, which refers to the repeated use of opioids leading to decreased analgesic efficacy, is a troubling problem in clinical pain treatment (Bobeck et al., 2019).The pathogenesis of opioid tolerance shares several characteristics with inflammatory pain. For instance, they are both characterized by opioid-associated hyperalgesia, and the mechanisms involve increased excitability of neurons in the dorsal horn of the spinal cord (central sensitization).The DRG exhibits neurochemical changes similar to inflammatory pain (Mao et al., 1995). Research has shown that AM is involved in the development of morphine tolerance.Coadministration of the selective AM receptor antagonist AM22-52 significantly attenuates the development of morphine tolerance (Hong et al., 2010). Additionally, blocking of the AM receptor prevents a decline in morphine efficacy and the related thermal hyperalgesia (Wang et al., 2011).Chronic morphine administration markedly increases AM expression in the superficial spinal cord and small-to-medium DRG neurons. A DRG explant culture study showed that chronic morphine application increased AM content and gene transcription in DRG neurons. Moreover, this upregulation of AM is μ-opioid receptor- and PKC signaling pathway-dependent (Hong et al., 2010). The mechanism of AM involvement in morphine tolerance may include a positive feedback process. Long-term use of morphine increases AM,which in turn increases its concentration through autocrine or paracrine mechanisms. Furthermore, intrathecal injection of AM receptor blockers inhibits the increase in CGRP in the spinal cord and DRG as well as the increase in nNOS in the spinal cord and the decrease of the endogenous opioid peptide BAM22 in the DRG (Wang et al., 2014c) induced by chronic morphine exposure. These findings suggest that another mechanism by which AM facilitates morphine tolerance involves increasing pain-inducing mediators and reducing pain-inhibiting molecules. In addition, inactivated AM inhibited the chronic morphine-evoked switch of the G protein of the G protein-coupled μ-opioid receptor from Gi to Gs in the spinal dorsal horn (Wang et al., 2016). Additionally, μ-opioid receptor desensitization is an important mechanism of morphine tolerance, which suggests that AM may be involved in the μ-opioid receptor desensitization induced by morphine tolerance. Further research found that blocking the AM receptor can inhibit the morphological changes in glia and the increase in the inflammatory factors interleukin-1 and interleukin-6 in the spinal cord induced by chronic morphine exposure (Zeng et al., 2014). AM is involved in morphine tolerance via activation of glia. In short, AM causes these neuronal and non-neuronal events to contribute to morphine tolerance.
One study found that upregulation of AM was induced by a single administration of morphine. Intrathecal combination with AM22-52 can increase the analgesic effect of morphine (Wang et al., 2014b). Therefore, a small amount of morphine causes an increase in AM activity.In vitroexperiments also confirmed that morphine at a certain dose increased AM content and release in neurons. This study suggested that the increased AM activity caused by chronic morphine exposure is actually the result of the accumulation of increased AM caused by each acute morphine administration. Each time morphine was administered, AM increased.The increased AM counteracts part of the analgesic effect of morphine. Eventually, AM accumulates and completely counteracts the effect of morphine (Huang and Hong, 2015).AM may antagonize morphine via two mechanisms. First,AM induces desensitization of the μ-opioid receptor, which reduces the morphine response. The other mechanism involves AM inducing an increase in other pain mediators,which increases the hyperexcitability of neurons. These two effects may occur in series or in parallel, leading to morphine tolerance.
Consistent with this inference, a laboratory study showed that chronic AM injection induced thermal/mechanical hypersensitivity and gradually reduced the analgesic effect of morphine (Wang et al., 2016; Huang et al., 2019a). The mechanism of this phenomenon is similar to that of chronic morphine exposure. For instance, chronic AM exposure switches the G protein coupled to the μ-opioid receptor in the spinal dorsal horn from Gi to Gs in a cAMP/PKA/CREB/ERK-dependent manner (Wang et al., 2016). Chronic AM induces activation of microglia and astrocytes and upregulates the expression of interleukin-1, interleukin-6,tumor necrosis factor (TNF)-α and nNOS in the spinal dorsal horn (Zeng et al., 2014). AM is also an important participant in peripheral sensitization. AM signaling mediates TNF-induced activities in the DRG (Chen et al., 2016).Inhibition of AM activity attenuates the TNF-α-induced upregulation of CCL-2, matrix metalloprotease 9 and CGRP.Meanwhile, chronic AM exposure increases the expression of glial fibrillary acidic protein and interleukin-1, which indicate enhanced activity of satellite glial cells. Blockade of AM receptors also inhibits TNF-α-induced activation of satellite cells. The CREB and nuclear factor-κB signaling pathway is involved in TNF-α and AM-induced activities (Chen et al., 2016). Chronic AM also increases the expression of TRPV1 in the DRG, a marker of pain hypersensitivity that is increased in pathological pain states (Huang et al., 2019a).Overall, these findings indicate that AM participates in or triggers upstream or downstream events that together can counteract morphine analgesia and accelerate morphine tolerance. This process could also be the mechanism by which AM participates in pathological pain. Inflammatory pain and cancer pain both cause increased expression of AM in key areas that are involved in pain information transmission.AM expression gradually increases during the development of neuropathic pain induced by spinal nerve ligation (unpublished data). Therefore, the series of cellular responses induced by chronic AM application is worth exploring.
AM not only mediates the development of morphine tolerance, but also participates in its maintenance because intrathecal injection of AM22-52 reverses or restores the analgesic effect of morphine. This action involves the ability of AM to recruit excitatory amino acids such as glutamate(Zeng et al., 2013). The mechanisms of morphine tolerance development and maintenance are not identical, but AM is involved in both, which could provide ideas for determining the mechanism and overcoming the clinical problem of morphine tolerance (Figure 2).
Adrenomedullin and Brain Injury
The role of AM in brain injury has been emphasized in recent years. Compared with its role in pathological pain (periphery or spinal cord injury), AM plays complex and controversial (positive or negative) roles in brain injury. Some clinical studies show that higher levels of AM correlate with a poor prognosis of stroke. For instance, circulating AM has been shown to be of prognostic significance in hemorrhagic or ischemic stroke. Enhanced plasma AM content has been associated with long-term outcomes of acute ischemic stroke (Somay et al., 2007; Zhang et al., 2014). The AM level in blood plasma is significantly increased in acute ischemic stroke patients. In the long-term clinical course, the AM level changes with the progression of neurological severity,infarct volume and functional prognosis (Serrano-Ponz et al., 2016). Blood AM levels on admission can predict outcomes after acute intracerebral hemorrhage (Wang et al.,2014a) and traumatic brain injury (Chen et al., 2014). Another study found that AM was increased in cerebrospinal fluid after traumatic brain injury (Robertson et al., 2001),and its mRNA expression was significantly increased in the ischemic cortex of an animal model of focal stroke produced by middle cerebral artery occlusion (Wang et al., 1995).Moreover, intracerebroventricular administration of AM exacerbated focal ischemic brain injury, which seems to show the pathogenic role of locally expressed AM in focal stroke(Wang et al., 1995).
Other than the abovementioned results indicating that AM aggravated brain injury, all other studies have found the opposite results. A previous study found that intravenous infusion of AM increases regional cerebral blood flow and prevents ischemic brain injury after middle cerebral artery occlusion by increasing the collateral circulation (Dogan et al., 1997). Another study found that one of the neuroprotective mechanisms of intravenous infusion of AM in reducing acute ischemic brain injury induced by transient middle cerebral artery occlusion may be inhibition of neutrophil infiltration (via myeloperoxidase) into ischemic tissue (Watanabe et al., 2001).
Continuous infusion of exogenous AM protects against ischemia/reperfusion injury by inhibiting the apoptosis of neuronal and glial cells and promoting angiogenesis by increasing NO formation and decreasing superoxide anion production (Xia et al., 2006). Intravenously administered AM reduces secondary injury and improves the outcome of animals with traumatic brain injury induced by fluid percussion (Gao et al., 2018). Serrano et al. (2002) found that AM expression in the cerebral cortex was altered after ischemic perfusion. AM was elevated in large pyramidal neurons and small neurons of all cortical layers as the reperfusion time increased in a reperfusion model induced by oxygen and glucose deprivation. Neurons that were not immunoreactive for AM were damaged, whereas neurons expressing AM were not affected (Serrano et al., 2002), suggesting a latent cell-specific protective role for AM. Several experiments were performed to confirm that circulating AM can defend neural cells against hypoxia-induced apoptosis via upregulation of Bcl-2 (Chaung et al., 2011; Wu et al., 2015) or inhibition of hypoxia-inducible factor-1 (Sun et al., 2014) in the nuclear factor-κB signaling pathway (Hu et al., 2015). Moreover, AM enhances cAMP levels and then activates PKA to protect neural cells from injury caused by hypoxia (Tokudome et al.,2002; Wang and Yang, 2009). AM has also been demonstrated to inhibit secretion of proinflammatory cytokines such as interleukin-1, interleukin-6 and TNF-α (Demir et al., 2013).In addition, AM was found to provoke endothelial PI3K/Akt activation, promote vascular regeneration and reduce the increase in endothelial permeability induced by cytokines,endotoxins or reactive oxygen species and thus may limit the formation of inflammatory exudates (Kim et al., 2002; Zhou et al., 2016). These findings suggested that AM can improve neurologic function, and its neuroprotective or repair effect is exerted via anti-apoptotic and anti-inflammatory effects.
The inconsistent findings regarding AM in brain injury as well as studies in genetically modified mice suggest that this issue is highly complex. For instance, when AM is knocked out in neurons in the brain, the infarct volume increases, and brain damage is aggravated in a permanent focal ischemia model. AM exerts a neuroprotective action in the brain that may be mediated by inducible NOS, matrix metalloprotease 9 and COX-2 (Hurtado et al., 2010). However, when AM is eliminated from endothelial cells, the infarct volume is reduced, and the animal exhibits less brain damage induced by middle cerebral artery occlusion. AM in the endothelium has an aggravating effect on brain injury by downregulation of the adhesion molecule Mcam and the proinflammatory mediator Lcn2 (Ochoa-Callejero et al., 2016). These results suggest that AM exerts different biological effects in various regions that independently or collectively participate in brain injury. These genetic experiments may provide an understanding of the conflicting results obtained by many laboratories worldwide.
Adrenomedullin and Regeneration in the Nervous System
Some progress has also been made regarding AM in the study of proliferation and differentiation of the nervous system. The application of AM for regulation of regeneration of various types of neuronal cells is a promising therapeutic strategy for the treatment of many neurodegenerative diseases. AM regulates the proportions different types of neurons,astrocytes and oligodendrocytes produced from progenitor cells (neural stem cells). AM-null animals exhibit obviously higher proportions of oligodendrocytes and lower proportions of astrocytes and neurons. Cells in which the AM gene is knocked out show tubulin hyperpolymerization and evident changes in the actin cytoskeleton, which are important processes in the formation and maintenance of mature neural cell morphology and physiology (Vergano-Vera et al.,2010; Martinez-Herrero et al., 2012).
Experimental evidence indicates that AM promotes oligodendrocyte precursor cell differentiation via an AM-receptor-PI3K/Akt pathway under conditions of pathological/hypoxic stress (Maki et al., 2015; Yanqin et al., 2017). Oligodendrocytes are glial cells that comprise the myelin sheath in the central nervous system. In particular, abnormal formation of the myelin sheath in white matter may lead to central nervous system disorders. Therefore, AM signaling may be a promising therapeutic method to promote oligodendrocyte regeneration in central nervous system diseases. Furthermore, AM also affects peripheral nerve regeneration, and AM and its receptors are highly enriched in primary cultures of Schwann cells and perineural fibroblasts. One study found that AM was released from perineural fibroblasts; the functional expression of AM receptors in perineural fibroblasts and Schwann cells was revealed by AM-induced stimulation of cAMP production. This finding suggests that local regulatory actions of AM are likely important for peripheral nerve regeneration (Dumont et al., 2002). Previous studies have shown that AM has a facilitatory effect on the reinnervation of perivascular nerves in phenol-injured perivascular nerve models (Hobara et al., 2007; Hashikawa-Hobara et al., 2012).AM is also involved in the proliferation of Schwann cell tumors, which is a hereditary disease involving malignant peripheral nerve sheath tumors (Hummel et al., 2010). The above evidence suggests a possible role of AM in nerve regeneration therapy.
Prospectives
Hyperexcitability of the neural network is a critical neurophysiological mechanism in some neurological disorders,including injury, sensory deprivation, epilepsy and tinnitus.Thus, the role of AM in neuropathic pain provides information on its role in other diseases. Furthermore, the role and mechanism of AM in brain injury reveals that AM may be involved in other neurodegenerative diseases such as Huntington's disease, multiple sclerosis, Parkinson's disease and Alzheimer's disease (AD). For instance, preliminary studies have found that AM plays a role in AD by showing that midregional proadrenomedullin was increased in mild cognitive impairment patients who were exhibiting the predementia stage of AD, and AM was also altered in patients with mild AD (Buerger et al., 2011; Fernandez et al., 2016). AM is also elevated in aging human brains, and mice lacking neuronal AM have better retention memory(Larrayoz et al., 2017). In addition, AM can cause cytoskeleton breakage by acting on cytoskeletal proteins, including tau and acetylated tubulin, and this process is involved in the loss of synaptic connections of the frontal cortex and hippocampus. AM enhances microglial activity and decreases neural cell adhesion molecule levels, cholinergic activity and neurotransmitters such as 5-hydroxytrytamine,dopamine and γ-aminobutyric acid, which are important in the mechanism of AD (Larrayoz et al., 2017; Ferrero et al.,2018). These results are consistently found and comprise preliminary evidence of the importance of AM in neurodegenerative diseases.
An important issue worth noting in the mechanism of AM involvement in pathological pain is glial cell activity. In addition to morphine tolerance, one study found that activation of satellite glial cells, microglia and astrocytes in the spinal cord is a crucial event in AM-involved pathological pain, including spinal nerve ligation-neuropathic pain and diabetic neuropathy. Additionally, AM can directly activate microglia and astrocytes via its receptors throughin vitropure culture of glial cells, and intraspinal injection of AM-activated astrocytes can cause long-term pain. These findings indicate that the activity of “immune cell-glial cell-neuron” networks provide insights into the mechanism of chronic pain. In fact, some studies have confirmed that glial cell activity is also a mechanism of brain injury. For instance, inhibition of inflammatory mediator release from microglia through the P2X receptor can be used to treat ischemic/hypoxic brain injury (Wang et al., 2013b). This information may also be used to clarify the mechanism of AM involvement in brain injury. In addition, the relationship between AM and NO is noteworthy. A recent study showed that AM activates its Gs-coupled receptor to increase cAMP levels and then promotes phosphorylation of endothelial NOS through the PKA pathway under the shear stress of blood flow (Iring et al., 2019). This is very similar to the mechanism of AM and NO in inflammatory pain and morphine tolerance. This phenomenon may occur in brain capillaries, neurons and glia in other neuropathological diseases. AM and CGRP belong to a homologous family. CGRP receptor antagonists are currently being developed to treat migraines and protect against brain injury. The role of AM in these two neuropathological conditions requires further study. In summary, a greater understanding and further exploration of AM in neurological diseases (Figure 3) is needed to provide a promising treatment strategy for these diseases.
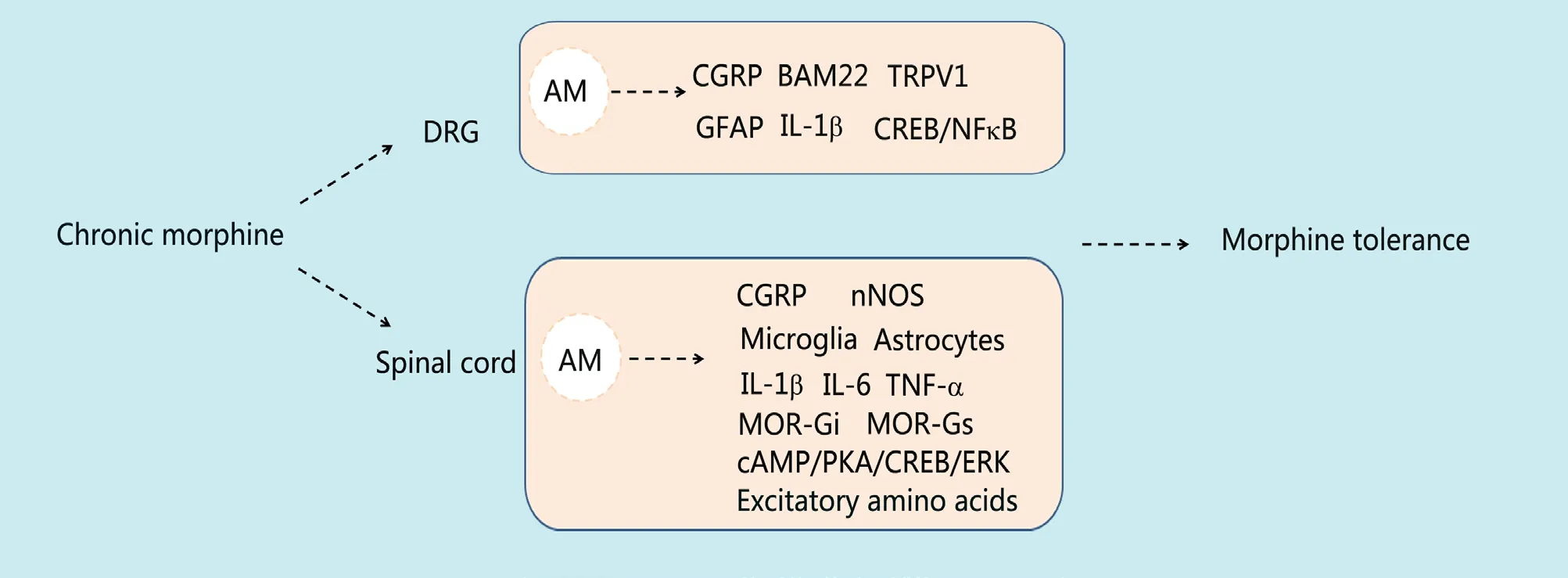
Figure 2 Summary schematic of the involvement of adrenomedullin in morphine tolerance.Chronic morphine administration increases the expression and release of AM in the DRG and spinal cord. AM increases pain-inducing substances in the DRG, such as CGRP, and decreases pain-suppressing substances, such as BAM22. Elevation of AM in the spinal cord activates microglia and astrocytes, alters MOR activity and promotes upregulation of other pain mediators such as CGRP. These activities induced by AM in the DRG and spinal cord increase the excitability of neurons and together promote the development and maintenance of morphine tolerance. AM: Adrenomedullin; BAM22: bovine adrenal medulla 22; cAMP: cyclic adenosine monophosphate; CGRP: calcitonin gene related peptide; DRG: dorsal root ganglia; ERK: extracellular signal-regulated kinase; GFAP: glial fibrillary acidic protein; IL: interleukin; MOR: μ-opioid receptor; NFκB: nuclear factor kappa B; nNOS: neuronal nitric oxide synthase; PKA: protein kinase A; TNF: tumor necrosis factor; TRPV1: transient receptor potential cation channel subfamily V member 1.
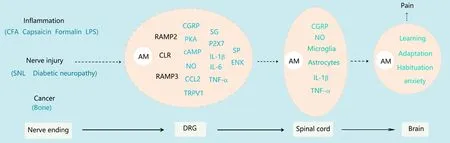
Figure 1 Summary schematic of the involvement of adrenomedullin in pathological pain.Peripheral noxious stimuli of different origins (inflammation, injury and cancer) are transmitted to the DRG, which induces an increase in AM expression. AM binds to its receptor elements to exert its activity, inducing the expression or release of a series of downstream factors in neurons and satellite glia, including CGRP, NO, CCL2 and TRPV1. These events together promote peripheral sensitization. Sensitized DRG neurons frequently emit action potentials that conduct to the central nervous system. The release of various substances, including AM, is enhanced from the central end of primary afferent fibers. AM can activate microglia and astrocytes to promote central sensitization and cause pathological pain. AM also plays a role in regulating brain processes such as learning and habituation. AM: Adrenomedullin; cAMP: cyclic adenosine monophosphate; CCL2:chemokine (C-C motif) ligand 2; CFA: complete Freund's adjuvant; CGRP: calcitonin gene related peptide; DRG: dorsal root ganglia; IL: interleukin; LPS: lipopolysaccharide; NO: nitric oxide; PKA: protein kinase A; SNL: spinal nerve ligation; TNF: tumor necrosis factor; TRPV1: transient receptor potential cation channel subfamily V member 1.
Author contributions:Literature search and manuscript writing: FJL,SRZ, and DMW. All authors approved the final version of the manuscript.
Conflicts of interest:The authors declare that there are no conflicts of interest associated with this manuscript.
Financial support:This work was supported by the National Natural Science Foundation of China, No. 81400922 (to DMW), 81571084; the Natural Science Foundation of Fujian Province of China, No. 2018J01813(to DMW); the College of Life Sciences of Fujian Normal University of China, No. FZSKG2018016 (to DMW).
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement:Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-Non-Commercial-ShareAlike 4.0 License, which allows others to remix, tweak,and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewer:Alfredo Martínez, CIBIR, Oncology, Spain.
Additional file:Open peer review report 1.
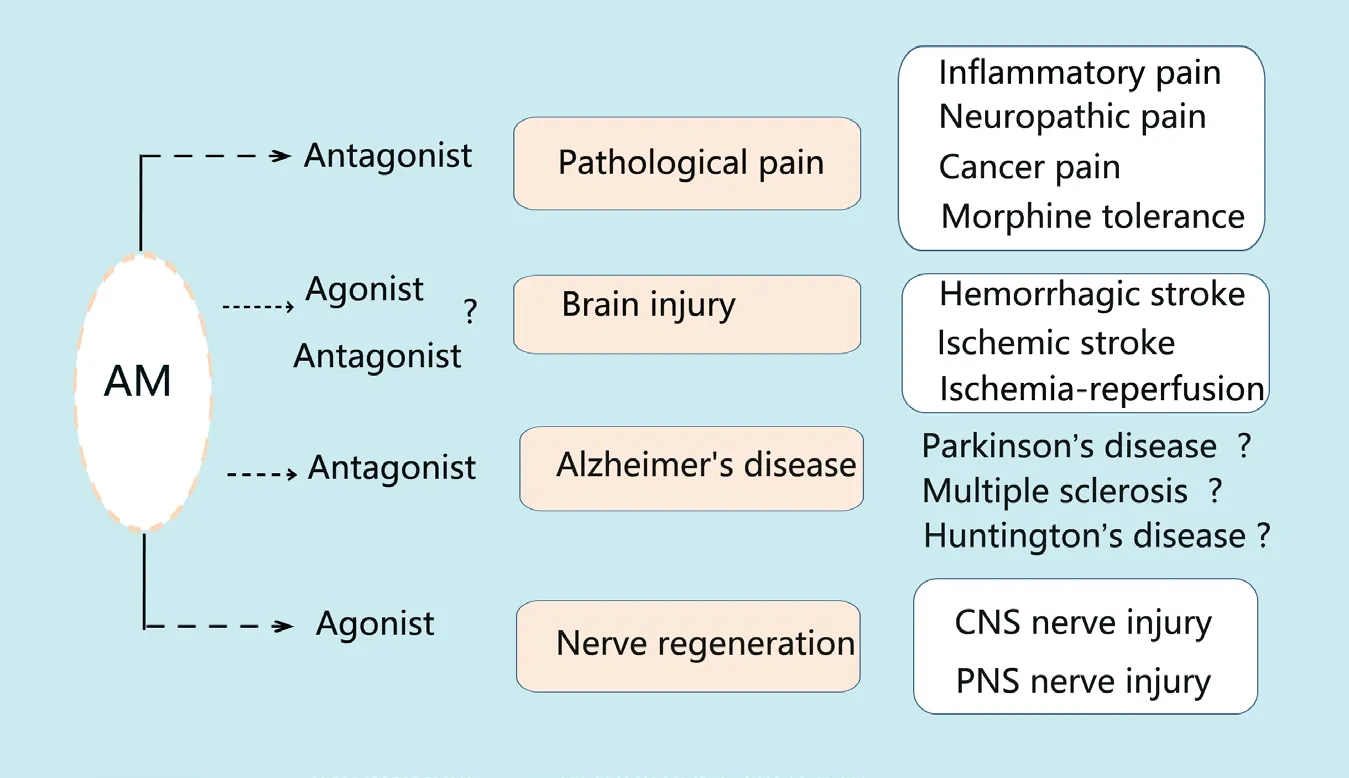
Figure 3 Summary schematic of potential applications of adrenomedullin for neuropathological diseases.Potential applications of AM for noxious pain(including inflammatory, neuropathic or cancer pain), morphine tolerance, brain injury, nerve regeneration and Alzheimer's disease are shown.Because AM plays different roles in various neuropathological diseases, the treatments differ and involve application of either AM antagonists or agonists. AM: Adrenomedullin; CNS: central nervous system; PNS: peripheral nervous system.
- 中國神經(jīng)再生研究(英文版)的其它文章
- Glial cells in intracerebral transplantation for Parkinson's disease
- Fast-tracking regenerative medicine for traumatic brain injury
- The N-formyl peptide receptors: contemporary roles in neuronal function and dysfunction
- Shifting equilibriums in Alzheimer's disease: the complex roles of microglia in neuroinflammation,neuronal survival and neurogenesis
- ABC efflux transporters at blood-central nervous system barriers and their implications for treating spinal cord disorders
- Biomaterials and neural regeneration