
The N-formyl peptide receptors: contemporary roles in neuronal function and dysfunction
2020-01-02 01:17PeterCussellMargaritaGomezEscaladaNathanielMiltonAndrewPaterson
中國神經(jīng)再生研究(英文版) 2020年7期
Peter J.G. Cussell, Margarita Gomez Escalada, Nathaniel G.N. Milton, Andrew W.J. Paterson
Centre for Biomedical Science Research, School of Clinical and Applied Sciences, Leeds Beckett University, Leeds, UK
Abstract N-formyl peptide receptors (FPRs) were first identified upon phagocytic leukocytes, but more than four decades of research has unearthed a plethora of non-myeloid roles for this receptor family. FPRs are expressed within neuronal tissues and markedly in the central nervous system, where FPR interactions with endogenous ligands have been implicated in the pathophysiology of several neurodegenerative diseases including Alzheimer's disease and Parkinson's disease, as well as neurological cancers such as neuroblastoma. Whilst the homeostatic function of FPRs in the nervous system is currently undefined, a variety of novel physiological roles for this receptor family in the neuronal context have been posited in both human and animal settings. Rapid developments in recent years have implicated FPRs in the process of neurogenesis and neuronal differentiation which, upon greater characterisation, could represent a novel pharmacological target for neuronal regeneration therapies that may be used in the treatment of brain/spinal cord injury, stroke and neurodegeneration. This review aims to summarize the recent progress made to determine the physiological role of FPRs in a neuronal setting, and to put forward a case for FPRs as a novel pharmacological target for conditions of the nervous system, and for their potential to open the door to novel neuronal regeneration therapies.
Key Words: Alzheimer's disease; formyl peptide receptor; neural regeneration; neuroblastoma; neurodegeneration;neuroinflammation; neuronal differentiation; stroke
Introduction
TheN-formyl peptide receptors (FPRs) are a family of G-protein-coupled chemoattractant receptors, and represent a critical modulator of host defence and inflammation (He and Ye, 2017). This receptor family is so-named because initial studies described receptor-mediated neutrophil chemotaxis activity, elicited by formylmethionine-containing peptides (Schiffman et al., 1975; Wilkinson, 1978). Original investigators postulated that the presence of a formylmethionine group was an imperative factor in ligand binding to FPR, and since the only natural sources of such peptides derive from bacterial or mitochondrial proteins, it was established that FPRs evolved to facilitate the recruitment of phagocytic leukocytes to loci of infection or tissue damage(Niedel et al., 1980). When it was discovered that formylated chemoattractant molecules, such as the archetypal cleavage product formyl-methionine-leucyl-phenylalenine (fMLF),could originate from both bacterial and endogenous mitochondrial sources, substantial evidence was provided for the endosymbiotic theory of the evolution of mitochondria from primitive bacteria (Margulis et al., 1985). This was further supported when endogenous formylated peptides emitted from the mitochondria of necrotic cells were shown to stimulate the recruitment of monocytes contributing to inflammatory responses through interaction with FPRs (Crouser et al., 2009). In addition to cell chemotaxis, Schiffman et al.(1975) reported that FPR activation by formylated peptides also induced the release of lysosomal enzymes by phagocytic leukocytes once at the pathologic site, further facilitating the clearance of invading pathogens and damaged tissue detritus. Subsequent researchers then demonstrated that myeloid FPR activation stimulated degranulation and the secretion of pro-inflammatory cytokines plus superoxide (Prossnitz and Ye, 1997; Le et al., 2002).
In more recent years, the cellular distribution and biological functions of the FPR receptor family have expanded to non-myeloid settings, where additional roles from organ homeostasis to acute inflammatory responses have been reported (He and Ye, 2017), demonstrating the adaptive nature of FPRs, and their distinct functionality relative to the cellular context in which they are expressed. Another crucial point of interest is the remarkably diverse ligand profile displayed by the FPR family; something for which FPRs have often been termed “promiscuous” (Migeotte et al., 2006). A plethora of structurally distinct endogenous FPR ligands have been identified, a number of which have been implicated in the pathophysiology of human diseases including HIV, amyloidosis, prion disease, stroke/ischemia reperfusion injury,stomach ulcers and several cancers (Li and Ye, 2013). While the role of FPRs in a host-defence setting has been extensively studied and well defined, recent research has highlighted high levels of FPR expression in the central nervous system(CNS) and peripheral nervous system (PNS) (Cattaneo et al.,2010), and although the biological significance of FPR expression in such a setting is currently undefined, a variety of novel physiological roles for FPRs in a neuronal context have been posited in both human and animal nervous system models. This review aims to summarize the recent progress made to determine the physiological role of FPRs in the neuronal setting, and to put forward a case highlighting FPRs as a novel pharmacological target for conditions of the nervous system, and for its potential to open the door to novel neuronal regeneration therapies (Figure 1).
Search Strategy and Selection Criteria
Searches were performed using PubMed encompassing literature published from 1975 to 31stAugust 2019. Eligibility criteria: reviews,in vivoandin vitrostudies, studies performed upon humans and animals and published in English.Key search words: ‘Formyl peptide receptor, Nervous, Neuronal, Neurodegeneration, Neuroinflammation, Alzheimer's disease, Parkinson's disease, Neurological cancer, Neuroblastoma, Glioblastoma, Neuronal differentiation, Neurogenesis,Neural regeneration.'
N-Formyl Peptide Receptor Family
In humans, three distinct FPR isoforms have been defined:FPR1, FPR2/ALX (formerly FPRL1) & FPR3 (formerly FPRL2), each encoded by a separate gene (FPR1,FPR2&FPR3, respectively), co-localized in a cluster upon the chromosomal region 19q13.3 (Ye et al., 2009). Attenuation of cellular responses to FPR agonists can be elicited by treatment with pertussis toxin, indicating that FPRs are coupled to the Giclass of G protein, as such FPR activation leads to transient calcium fluxes, ERK phosphorylation and cell locomotion (Suzuki et al., 1996; Wenzel-Seifert et al., 1999). Ligands for FPRs have also been suggested to have the potential to act as biased agonists and therefore the assigning of activation pathways to individual agonists and receptors is more complex (Raabe et al., 2019). FPR1 was the first isoform to be identified as the binding site for N-formylated peptides such as the prototypic chemoattractant fMLF, which FPR1 binds with high affinity (KD<1 nM) (Ye et al., 2009). FPR2 shares a 69% sequence identity with FPR1, yet has been defined as a low affinity fMLF receptor; although FPR2 has shown to become activated via fMLF at micromolar concentrationsin vitro(Prossnitz and Ye, 1997), it remains unclear as to whether such high concentrations of formylated peptide would exist in a natural setting, and therefore it is yet to be determined as to fMLF representing the true endogenous ligand of FPR2 in humans. FPR3 shares a 58% and 83%sequence identity with FPR1 and FPR2 respectively, but is unable to bind formyl peptides (Migeotte et al., 2005). The F2L peptide identified as an endogenous peptide ligand for FPR3 is derived from the human intracellular heme-binding protein 1 (Migeotte et al., 2005) and it's generation involves cathepsin D mediated cleavage (Devosse et al., 2011). Rabiet et al. (2011) studied the expression pattern of FPR3 in human embryonic kidney cells, and found FPR3 to display a marked level of resting phosphorylation versus other FPR isoforms. Furthermore, FPR3 was found to be expressed within small intracellular vesicles despite a lack of agonist stimulation, suggesting that unlike resting FPR1/2 which localise in the plasma membrane, FPR3 exhibits some level of constitutive endocytosis. In addition to this, Rabiet et al.(2011) reported that FPR3-expressing cells underwent no G-protein activation, indicating that rather than transducing signal, FPR3 could serve some “l(fā)igand scavenging” function similar to that displayed by chemokine receptors D6 and ACKR3. Based upon these observations, it was posited, and today broadly accepted that FPR3 serves contrasting physiological functionality to that of FPR1 and FPR2. Notwithstanding, the nature and mechanism of FPR3 function is poorly understood at present. Recent studies have indicated that the FPRs can both undergo homo- and hetero-oligomerization leading to greater diversity in the receptor population and signaling repsonses (Cooray et al, 2013).
Single nucleotide polymorphisms have been identified in FPR receptor genes and have been suggested to have disease modifying effects (Miettinen, 2011; Liang et al., 2014; Zhang et al., 2017a). Links between these and macular degeneration have been suggested (Liang et al., 2014), however, specific links to the neurological disorders Alzheimer's disease (AD)and Parkinson's disease (PD) plus neurological cancers remain to be fully identified and characterized.
The history of FPRs from an evolutionary perspective is a complex one, and is thought to have been driven by positive selection leading to functional diversification (Muto et al.,2015), despite conserving a high level of sequence homology across species, the number of FPR family members varies significantly from one genome to another; for example the mouse FPR (mFPR) model contains eight identified mFPR coding genes (Gao et al., 1998)versusjust three in humans(Ye et al., 2009). He et al. (2013) studied the mouse homologs of FPR1, FPR2 and FPR3 and demonstrated only slight differences in agonist preference, binding properties and cellular distribution relative to the human equivalents; and therefore ligands with specificity for human FPR1/FPR2 generally display specificity for the relative murine ortholog.In relation to the additional mFPRs, many (mFPRs 4-7)have been identified within olfactory sensory neurons and the murine vomeronasal organ, it is known that mice rely on olfaction for communication and environmental feedback,and Riviere et al. (2009) speculates that mouse FPRs are a powerful sensory tool through which mice can detect contaminated compounds, or via the assessment of secretions,facilitate the identification of unhealthy conspecifics. This large scope for variation in FPR family members and endogenous ligands between species highlights the need for care when interpreting animal research data in relation to human extrapolation.
FPRs are well known for their promiscuity, and have shown to bind a vast number of structurally distinct ligands including endogenous and bacterial derived peptides, synthetic library peptides, small non-peptide molecules and lipids. This unusual diversity of ligands has led to the classification of FPRs as pattern recognition receptors (Li et al.,2016). In particular, FPR2 displays an astonishing variation in its ligand profile, and since it was demonstrated as a high affinity receptor for the eicosanoid mediator Lipoxin A4(Maddox et al., 1997), FPR2 represents the only identified chemotactic G-protein-coupled chemoattractant receptor that can be activated by both endogenous peptide and lipid ligands. Although FPRs were initially identified in myeloid cell populations, immunocytochemical studies since undertaken revealed broad FPR expression throughout a variety of cell types and tissues. In particular, studies on human and mammalian models have detected FPR1, FPR2 and FPR3 mRNA and/or protein in a range of CNS and peripheral nervous system tissues including; amygdala, autonomic nervous system (both branches), basal ganglia, cerebellum, cerebral cortex, corpus callosum, hippocampus, hypothalamus,medulla, midbrain, olfactory region, pituitary gland, pons,retina, sensory system, spinal cord, striatum and thalamus(Becker et al., 1997; Migeotte et al., 2006; Cattaneo et al.,2010; Ho et al., 2018). FPRs are also expressed in vascular tissue which would allow FPR ligands to have effects on the brain via the blood supply. One can begin to shed light on the biological significance of FPR expression in the nervous system by examining some FPR interactions with endogenous compounds which have been implicated in the pathogenesis of several neurological disease states.
N-Formyl Peptide Receptor and Neurodegeneration/Stroke
Neurodegenerative disorders such as AD and PD, are often exacerbated by pro-inflammatory activity, through which neuroinflammation is brought about via chronic activation of glial cells including microglia and astrocytes (Mollica et al., 2012). Although the vast majority of CNS cells descend from a common neuroectodermal/neuroepithelial progenitor, microglial cells descend from myeloid lineage, and when functioning normally act as the front line of protection in the brain from infection or injury (Ransohoff et al., 2010).Once in a hyperactive state however, glial cells secrete elevated levels of reactive oxygen species (ROS) and inflammatory molecules, which can lead to neurotoxicity. In turn, this neuronal damage can lead to further recruitment and activation of glial cells, and thus additional neuronal stress (Ndubaku et al., 2008). Herein lies the cyclical nature of chronic neuroinflammatory conditions, and interestingly there exists a close relationship between chemoattractant receptors like FPRs and neuroinflammation. The significant contribution of inflammatory signaling events in such disease models has led to novel therapeutic attempts to treat neurodegeneration by attenuating inflammation.
AD is a neurodegenerative disorder characterized by intracellular neurofibrillary tangles derived from hyperphosphorylated tau protein and aggregates of the amyloid-β(Aβ) peptide in extracellular plaques. The Aβ peptide is a cleavage product of the amyloid precursor protein that plays a key role in producing chronic neurotoxicity and it's actions have been suggested to be in part receptor mediated (Jarosz-Griffiths et al., 2016; Mroczko et al., 2018). Interestingly,FPR2 has demonstrated to be a high-affinity receptor for the 42-amino acid form of Aβ. Through binding and activating FPR2 present in the microglial cell membrane (Yu and Ye,2015), Aβ has demonstrated to stimulate the recruitment and activation of microglial cells and mononuclear phagocytes from the blood supply. Moreover, much like that observed in peripheral macrophages during immune responses, once at the pathologic site, FPR-activated microglia secrete complement proteins, pro-inflammatory cytokines and ROS, which over time can lead to gliosis and neurotoxicity (Le et al., 2001; Slowik et al., 2012). The neurotoxic actions of Aβ have also been suggested to involve ROS and in particular hydrogen peroxide (Milton, 2004), which could be mediated via activation of FPR2. Furthermore, dense populations of microglia frequently surround the Aβ plaques observed in models of AD, and this, coupled with the detection of upregulated neurotoxic mediators led some to believe that the pathology of AD was caused by inflammation and microglial activation (Rogers et al., 2007). It is entirely plausible that FPR2/Aβ interactions may be a key mechanism behind this widely reported microglial recruitment, plus the release of inflammatory and neurotoxic mediators observed in AD;in vitrostudies upon munrine neutrophils revealed that Aβ induces both chemotaxis and oxidant production via FPR2, suggesting the presence of a combined molecular basis for microglial recruitment and initiation of oxidative stress in AD (Tiffany et al., 2001). Another important role of FPR2 in AD was highlighted by Yazawa et al. (2001), who demonstrated that upon incubation with macrophages, Aβ-FPR2 complexes rapidly internalized into the cytoplasmic compartment. While transient FPR2 activation by Aβ stimulated rapid degradation of the protein, chronic stimulation produced a build-up of Aβ-FPR2 complexes leading to the formation of fibrillar aggregates. If we consider once more the AD plaque; tightly surrounded by activated microglia and with an abundant source of Aβ, it could be possible that FPR2 represents a means through which Aβ is internalized by said microglia, stimulating the formation of the destructive senile plaques augmenting AD pathology. The findings of Yazawa et al. (2001) are significant, as it highlights FPR2 as not only mediating pro-inflammatory pathophysiological effects, but also the internalization of Aβ itself. What's more,it displays a clear link between neuroinflammation and Aβ plaque formation. Another significant theory implicating FPRs in AD neuroinflammation was put forward by Wilkins et al. (2015), who proposed that damage associated molecular pattern (DAMP) molecules, such as formylated peptides emitted from damaged mitochondria, could directly contribute to AD neuroinflammation. Following treatment of cultured neuronal and microglial cells with mitochondrial lysates, increased levels of inflammatory cytokines such as tumor necrosis factor α (TNFα) and interleukin (IL)-8 were detected. This demonstrates that mitochondrial DAMPs,including FPR ligands, can themselves directly induce AD-like neuroinflammatory activity. Could these molecules be bringing about this pro-inflammatory activity via FPR signaling? These studies have contributed to our understanding of AD pathology, and implicate FPRs as a significant mediator. Nevertheless, one key “chicken-and-egg” question remains: is neuroinflammation via FPR2/Aβ or FPR/DAMP interactions the cause of AD aetiology, or merely a response to some other initial insult?
Activated microglia have been implicated not only in AD,but in the pathology of various disorders of the CNS, including PD. PD is characterized by the degradation of dopaminergic neurons in the region of the substantia nigra and the development of intraneural Lewy bodies. and when large numbers of activated microglia were reported in the substantia nigra of PD patients (McGeer et al., 1988), researchers posited that microglia may stimulate the degradation of dopaminergic neurons contributing to PD pathology. This was substantiated further when pro-inflammatory cytokines such as IL-1β and IL-6 (Blum-Degen et al., 1995), as well as TNFα(Mogi et al., 1994) were identified in the brain and cerebrospinal fluid of PD patients. Pro-inflammatory cytokines such as these have been widely shown to be released by microglia in direct response to FPR activation by endogenous inflammatory mediators (Mollica et al., 2012) so, like in AD, could microglial activation by FPR activity once again play a pivotal role in PD? The prodromal phase of PD is markedly long and can regularly surpass ten years. During this preclinical phase,various non-motor symptoms regularly present including hyposmia, anxiety, sleeping difficulties and sundry gastrointestinal disturbances from gastroparesis to severe constipation (Postuma, 2019). Interestingly, Lewy bodies have been detected in neurons of the enteric nervous system (ENS) and vagus nerve of PD patients at this early prodromal stage, mirroring the latter stage neuropathology observed in the CNS(Pouclet et al., 2012; Cersosimo et al., 2013). This has led many to believe the gastrointestinal tract to be the epicentre of PD pathogenesis, and in one hypothesis, a combination of mitochondrial dysfunction and a dysbiotic gut microbiome is proposed to trigger a cascade of events whereby DAMPs such as formylated peptides trigger neuronal innate immunity in the ENS culminating in neurodegeneration (Cardoso and Empadinhas, 2018). The bacterial origin of mitochondria here becomes significant; upon exposure to a foreign pathogen, the innate immune system learns to recognize not a specific pathogen, but instead learns to recognize certain conserved features known as pathogen associated molecular patterns. The most commonly exploited pathogen associated molecular pattern that is conserved in both mitochondrial and bacterial proteins is the presence of a formylmethionine group, and as such damaged mitochondria can trigger innate immunity via FPR activation (Le et al., 2002). Mitochondria mediate numerous immunohomeostatic functions, and many bacteria have developed mechanisms which specifically target host mitochondria with the release of toxic factors. It has therefore been proposed that gut dysbiosis, which is increasingly reported in early stage PD, promotes the release of inflammatory factors including formyl peptides plus microbial toxins which target mitochondria of ENS neurons, triggering microglial activation and subsequent ENS neuroinflammation, that in turn progresses from the gut to the brain via the vagus nerve (Cardoso and Empadinhas, 2018). Remarkably, it is therefore plausible that PD could be triggered by some endogenous gut pathogen which acts directly upon FPRs. This theory of PD pathology has been reinforced byin vivostudy data: the administration of bacterial metabolites to germ-free transgenic mice provoked PD-like symptoms (Sampson et al.,2016), and patients who underwent a full truncal vagotomy were found to have a significantly decreased risk of developing PD, indicating that this is the most likely passage of the disease from gut to brain.
It is important to note that FPR activation stimulates not only pro-inflammatory activity, but in fact upon binding an exclusive group of endogenous ligands, FPRs have shown to stimulate pro-resolving anti-inflammatory activity. For example, via FPR2, the protein and lipid mediators Annexin A1 and Lipoxin A4 have shown to produce neutrophil apoptosis and macrophage efferocytosis, and knockout FPR2 mice have shown to lack the ability to resolve inflammation(Dufton et al., 2010), Annexin A1 has shown to resolve inflammation in the brain during sepsis (Gavins et al., 2012),and has also shown to stimulate Aβ degradation (Reis et al., 2016) all through FPR2 signaling. Meanwhile another inflammatory mediator, serum amyloid A (SAA) has shown to stimulate both beneficial and destructive inflammatory processes through FPR2 interaction; monocytes produce pro-inflammatory cytokines such as TNFα in response to low concentrations of SAA, yet in response to high SAA concentrations, have been shown to produce the inflammation-resolving cytokines like IL-10 (Lee et al., 2006). The administration of Resolvin E1, another agonist of FPR2, in combination with Lipoxin A4 resolved inflammation in a mouse model of AD (Kantarci et al., 2018). Another of the resolvins, Resolvin D1 was demonstrated to halt remote neuroinflammation and stimulate neuroprotective effects leading to functional recovery following acute focal brain damage (Bisiccia et al., 2018). This ability of FPR2 to transduce both pro- and anti- inflammatory activity may suggest multiple binding sites exploited by different agonists (Le et al., 2005). Cooray et al. (2013) suggested that this shift in FPR2-mediated pro- and anti-inflammatory cell responses comes about via conformational changes of the receptor upon ligand binding: it was shown that binding of anti-inflammatory ligands like Annexin A1 triggered FPRs to form homodimers, leading to the release of inflammation-resolving cytokines like IL-10. On the other hand inflammatory ligands like SAA did not cause receptor homodimerization.
These pro-resolving actions of FPR ligands and receptors are very relevant to stroke therapy. The FPR2 receptor has been implicated in platelet function which is also highly relevant to stroke (Vital et al., 2016; Senchenkova et al., 2019).
AD and PD are just two examples of CNS disease in which FPR interactions with endogenous/exogenous ligands can bring about a state of neuroinflammation by chronic activation of microglia and of neurotoxicity via the release of neurotoxic factors. Through similar mechanisms, FPR activity has also been implicated in the progression of various other diseases of the CNS, among them amyotrophic lateral sclerosis (Zhang et al., 2011), bacterial meningitis (Braun et al., 2011), and prion disease (Le et al., 2001). As such, FPRs should represent a novel therapeutic target for future drug development in this area. In these disease states however,it remains unclear as to when microglial recruitment and subsequent activation first occurs during disease pathology.Nevertheless, if FPR-mediated inflammatory signaling were to be pharmacologically overcome, it would unquestionably reduce neurodegenerative disease progression.
N-Formyl Peptide Receptors and Neurological Cancer
Recent studies have highlighted the involvement of FPRs in the progression of several neurological cancers. In numerous cases, primary tumors have been found to exploit FPR regulation in order to escalate growth: neuroblastoma primary tumors and cell lines have been demonstrated to express FPR1, and increased FPR1 tumor expression is correlated with high-risk disease and low survival rates relative to low FPR1-expressing tumors (Snapkov et al., 2016). Knockdown of FPR1 with shRNA delays neuroblastoma development,while ectopic overexpression of FPR1 elicits augmented tumorigenesis in nude mice (Snapkov et al., 2016). This demonstrates functional FPR activity in human neuroblastoma cells, and FPR overexpression resulted in a markedly increased tumor load, suggesting that FPR1 is implicated in producing an aggressive neuroblastoma phenotype. FPRs have also shown to be expressed by human glioblastoma cell lines, in which it has been suggested that FPR activation exacerbates tumour malignancy through the production of angiogenic factors and the activation of epidermal growth factor (Huang et al., 2008). Highly malignant human glioblastomas have been reported to selectively overexpress FPR1; its activation promoting cancer progression and metastasis (Zhou et al., 2005). FPR activity aiding cancer progression via increasing angiogenesis and metastasis appears to be a common theme in many neurological cancers, and this is often brought about via interactions with endogenous agonists such as Annexin A1, which was found to exist in high concentrations in xenografts of human neuroblastoma raised in nude mice, and knockdown of FPR and Annexin A1 reduced tumour growth significantly (Yang et al., 2011).
Activation of FPR1 in human astrocytoma cell lines promotes motility, growth and angiogenesis. Targeting FPR1 with a specific antagonist was found to reduce astrocytoma cell motility and activation, thus prolonging the survival of tumour-bearing mice (Boer et al., 2013). These studies demonstrate that the stimulation of FPR in neurological cancer cells leads to FPR upregulation in order to increase cell proliferation and tumor growth in an autocrine or paracrine manner. However, discrete changes in FPR modulation can lead to a range of biological responses depending on the cellular context, and as such FPR upregulation has been shown to produce stimulatory and inhibitory effects upon tumor progression depending on the cancer histotype (Prevete et al., 2015). Combined, these studies may suggest that targeting FPR1 may be beneficial in the treatment of the aforementioned neurological cancer states, although pharmacological interventions would have to be highly specified to target the area of pathology, as successful adjuvant chemotherapy may require functional FPR1 (Weiss et al., 2018). Baracco et al.(2016) found that specific FPR1 antagonism reduced the effi-cacy of chemotherapy in a mouse breast cancer model via an immmunosupressive action, whilst this is not in a neuronal setting it may suggest that the multiple actions of FPR could play a rolein vivoand limit the effectiveness of FPR1 antagonism in cancer chemotherapy.
N-Formyl Peptide Receptors in Neural Regeneration
While the discovery of FPR interactions associated with neurological disease progression spiked much interest into FPRs as a novel pharmacological target to resolve such conditions,recent years have seen a marked number of studies focusing upon determining the physiological function of FPRs in the context of a healthy nervous system. Some remarkable findings have been unearthed, such as the implication of FPR transduction in the process of neurogenesis, which could represent a significant development in the field of neuronal regeneration in the future. Wang et al. (2016) demonstrated that fMLF induces the differentiation of murine neural stem cells (NSCs) into neurons, confirmed by elevated levels of neuronal markers DCX24 and TUJ1. In a further investigation by this team, Zhang et al. (2017b) reported that during differentiation initiated by fMLF, NSCs increased the expression of both FPR1 and FPR2, and while FPR activation promoted differentiation of NSCs into neurons,their differentiation into astrocytes and oligodendrocytes was simultaneously inhibited. Furthermore, NSC neuronal differentiation was shown to be mediated via P13K-AKT signaling events leading to ROS generation. These findings posit a functional role of FPR in neurogenesis, and it may be possible to exploit this mechanism for NSCs to target areas of inflammation or damage in the nervous system; following nervous trauma, such as that elicited by cerebral ischemia,inflammatory mediators are released, promoting the migration and differentiation of NSCs into neurons. Both FPR1 and FPR2 are present in NSCs, and FPR activity has shown to mediate NSC chemotaxis bothin vitro&in vivo. Hence,FPR activation by specific agonists could manipulate this axis to target areas of nervous system damage and regenerate damaged neuronal tissue. While promising, these studies only focus upon murine NSCs, and in light of the differences in FPR family members and ligand preferences between species (Ye et al., 2009), the same outcomes/mechanisms of transduction may not necessarily extrapolate to the human situation.
In vitrodata for FPR-mediated neuronal cell differentiation including both murine and human neuronal cells was provided by Cussell et al. (2019), who reported that the synthetic nonpeptide FPR agonist FPRa14 induces neuronal cell differentiation of mouse and human neuroblastoma cells. FPRa14 stimulated novel differentiated cell phenotypes which were distinct from one another, and presented individually in a dose-dependent manner. These novel differentiated cells were demonstrated in murine Neuro2a, human IMR32 and human SH-SY5Y cell lines (Figure 2). Additionally, in murine Neuro2a cells siRNA knockdown of FPR1 and FPR2 attenuated neuronal differentiation indicating that both FPR1 and FPR2 are required in order to elicit neuronal differentiation into these novel forms. FPR activity has also been implicated in the differentiation of glia following nerve injury. Schwannoma cells exposed to fMLFin vivoproduced axon-like processes (Korimová et al., 2018), a phenomenon which was blocked via preincubation with PBP10; a selective antagonist of FPR2, suggesting that DAMPs such as fMLF emitted from axonal mitochondria may trigger Schwann cell differentiation in order to aid neuronal regeneration following nervous system trauma. Additionally, immunohistochemistry revealed marked FPR2 localization within the growth cones, indicating that FPR2 could play a key role in axonal genesis and outgrowth (Korimová et al., 2018). This theory is reinforced by the findings of Ho et al. (2018), who reported that treatment of primary hippocampal neurons with FPR antagonists WRW4 and PBP10 significantly reduced their natural axonal and dendritic outgrowth. These studies provide evidence for FPRs having a physiological role in the nervous system, and together posit FPRs as a viable target for neuronal regeneration therapies that may be used in the treatment of brain/spinal cord injury, stroke and neurodegeneration.
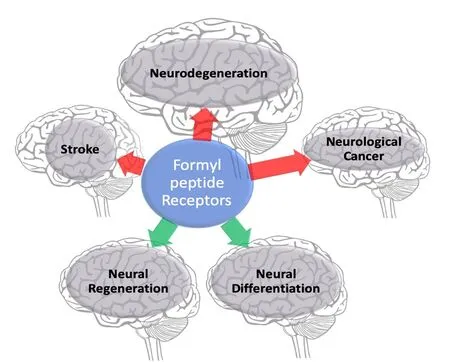
Figure 1 Summary of published formyl peptide receptor implications in neurological disorders.
If FPR-mediated neuronal differentiation were to be better characterized, another potential application could come within the field of neuropathic pain. Injury to peripheral nerves can present following trauma, surgery and amputation, and often result in the formation of painful neuromas;masses of hypersensitive nervous tissue driven by uncontrolled neuronal differentiation leading to blind-ended axons and proliferating connective tissue (Black et al., 2008). Could this neuroma formation be driven by FPR signaling? Following trauma to the nervous system, DAMPs like fLMF are emitted from damaged mitochondria. If these interact with FPRs present in the local nervous tissue, this could initiate neuronal differentiation leading to uncontrolled axonal and dendritic outgrowth, ultimately presenting as a neuroma. In such a case it may be possible to administer FPR antagonists in order to curtail this undesirable neuronal differentiation,and potentially halt neuroma formation, preventing neuropathic pain. On the other hand FPR agonists have also been correlated with inhibitory effects upon neuropathic pain.Following treatment with Lipoxin A4, CD1 mice and Wistar rats that underwent spinal hemisection at T10 were found to have significant reductions in the intensity of mechanical pain hypersensitivity and spinal expression levels of microglial markers and pro-inflammatory cytokines induced by SCI, when compared to rodents receiving control vehicle injections (Martini et al., 2016). This suggests a dual anti-inflammatory and analgesic action of the endogenous lipid moderator when interacting with FPR2. For FPR treatments of neuropathic pain conditions to be effective, the role of FPRs in neuronal regeneration following injury should be further explored.
Future Possibilities in Nervous System N-Formyl Peptide Receptor Research

Figure 2 Summary of findings by Cussell et al. (2019) demonstrating morphological effects of FPRa14 upon neuroblastoma cell lines and the ability of FPR1-specific antagonists Boc-MLF and Cyclosporin H to attenuate N-formyl peptide receptor (FPR)differentiation.
There is a substantial interest at present to identify novel protein targets and biomarkers of neurological diseases, and the FPRs or their ligands represent potential candidates. The association of FPR transduction with neuronal cancers and neurodegenerative disease states has been reported extensively. FPR signaling has shown to progress disease pathology both via direct interaction with neuronal cells and in an indirect manner via inflammatory mediators produced by FPR-activated microglia. The neuroinflammatory contribution of FPRs to such conditions represents a key mechanism to overcome in order to attenuate disease progression. While anti-inflammatory interventions seem to be an evident route into resolving neuroinflammation; clinical trials attempting to resolve neuroinflammation in AD patients with generic anti-inflammatory drugs have had limited success [reviewed in Zhu et al. (2018)]. This may be due to the window of treatment being too short to take significant effect, or because the intervention was administered once the pathology had progressed too far. It may however be beneficial to instead consider the application of FPR-specific pro-resolving lipid mediators Lipoxin A4 or Resolvin D1; via FPR2 these have a potential dual action in diminishing microglial recruitment,and reducing the production of pro-inflammatory or neurotixic factors. However FPR2 ligands have been suggested to have neurotoxic properties, for example FPRa14 toxicity is blocked by FPR2 specific antagonists and siRNA (Cussell et al., 2019). Targeting FPR directly with careful choice and testing of actions may prove more efficacious versus generic anti-inflammatory interventions.
The recent implication of FPRs in the process of neurogenesis and neuronal differentiation are significant, and represent a novel target for neuronal regeneration therapies.Despite promising developments fromin vitrostudies, there has, at present, been very little animal experimentation targeting FPR for neuronal regeneration, and this should be considered in the near future. Therefore any FPR compound used in this way should be highly specific for the locus of interest to minimize potential contra-indications and this represents a key challenge to overcome when developing potential FPR-based neuronal regeneration interventions. Whilst there are many vital homeostatic FPR interactions that occur throughout the body, FPR knockout mice are viable and no neuronal deficits have been reported to date indicating a need for more experimentation exploring the physiological actions of FPR in the nervous system.
This review has outlined compelling evidence suggesting FPRs playing physiological roles in the nervous system, and highlighted the actions of FPR signaling upon neurological disease states. However before this complex receptor family should be considered as a pharmacological target for conditions such as AD, gaining a better understanding of basic FPR functionaltily in a nervous setting is first important in order to inform potential drug development in the future.Recent reports of FPR activity eliciting neuronal differentiation and neurogenesis are promising for the field of neuroregeneration, but at present largely have relied onin vitromodels. Targetting FPR therefore has enormous potential,and once bolstered with a greater understanding of the receptor family through research, may bring about long-awaited options in the treat treatment of brain/spinal cord injury,neurological cancer, stroke and neurodegeneration.
Author contributions:PJGC, MGE, NGNM and AWJP wrote and critically reviewed the manuscript. All authors approved the final manuscript.
Conflicts of interest:The authors declare the following competing financial interest(s): NGNM is named as the inventor on a UK patent held bythe University of Roehampton for the use of kissorphin peptides to treat Alzheimer's disease, Creutzfeldt-Jakob disease or diabetes mellitus (Publication Numbers: GB2493313 B); under the University of Roehampton rules he could benefit financially if the patent is commercially exploited.NGNM is also a shareholder and director of NeuroDelta Ltd (Company No. 06222473; http://www.neuro-delta.com).
Financial support:This work did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors. PJGC was supported by a Leeds Beckett University PhD studentship.
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-Non-Commercial-ShareAlike 4.0 License, which allows others to remix, tweak,and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewers:Farid Menaa, California Innovations Corporation, USA; Baruh Polis, Bar-Ilan University, Israel.
Additional file:Open peer review reports 1 and 2.
- 中國神經(jīng)再生研究(英文版)的其它文章
- Glial cells in intracerebral transplantation for Parkinson's disease
- Fast-tracking regenerative medicine for traumatic brain injury
- Adrenomedullin: an important participant in neurological diseases
- Shifting equilibriums in Alzheimer's disease: the complex roles of microglia in neuroinflammation,neuronal survival and neurogenesis
- ABC efflux transporters at blood-central nervous system barriers and their implications for treating spinal cord disorders
- Biomaterials and neural regeneration