
Implication of a novel truncating mutation in titin as a cause of autosomal dominant left ventricular noncompaction
2022-05-06 08:32XueQiDONGDiZHANGYiQUYuXiaoHUChunXueYANGTaoTIANNanXUHaiLunJIANGLiZENGPengYanXIAYaXinLIURuiLIUXianLiangZHOU
Journal of Geriatric Cardiology 2022年4期
Xue-Qi DONG, Di ZHANG, Yi QU, Yu-Xiao HU, Chun-Xue YANG, Tao TIAN, Nan XU,Hai-Lun JIANG, Li ZENG, Peng-Yan XIA, Ya-Xin LIU,?, Rui LIU,?, Xian-Liang ZHOU,?
1. Department of Cardiology, Fuwai Hospital, National Center for Cardiovascular Disease, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China; 2. Department of Echocardiography, Fuwai Hospital, National Center for Cardiovascular Disease, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing,China; 3. Institute of Medicinal Biotechnology, Chinese Academy of Medical Sciences and Peking Union Medical College,Beijing, China; 4. State Key Laboratory of Membrane Biology, Institute of Zoology, Chinese Academy of Science, Beijing,China
ABSTRACT BACKGROUND Mutation in the titin gene (TTN) in left ventricular noncompaction (LVNC) has been reported with a highly heterogeneous prevalence, and the molecular mechanisms underlying the pathogenesis of TTN gene mutation are uncharacterized. In the present study, we identified a novel TTN mutation in a pedigree with LVNC and investigated the potential pathogenic mechanism by functional studies. METHODS The whole-genome sequencing with linkage analysis was performed in a 3-generation family affected by autosomal dominant LVNC cardiomyopathy. The clustered regularly interspaced short palindromic repeats associated protein 9 (CRISPR/Cas9) technology was used to establish novel truncating mutation in TTN in a rat cardiomyoblast H9C2 cell line in vitro, in which functional studies were carried out and characterized in comparison to its wild-type counterpart. RESULTS A novel truncating mutation TTN p. R2021X was identified as the only plausible disease-causing variant that segregated with disease among the five surviving affected individuals, with an interrogation of the entire genome excluding other potential causes. Quantitative reverse transcription-polymerase chain reaction and cellular immunofluorescence supported a haploinsufficient disease mechanism in titin truncation mutation cardiomyocytes. Further functional studies suggested mitochondrial abnormities in the presence of mutation, including decreased oxygen consumption rate, reduced adenosine triphosphate production, impaired activity of electron translation chain, and abnormal mitochondrial structure on electron microscopy. Impaired autophagy under electron microscopy accompanied with activation of the Akt-mTORC1 signaling pathway was observed in TTN p.R2021X truncation mutation cardiomyocytes. CONCLUSIONS The TTN p. R2021X mutation has a function in the cause of a highly penetrant familial LVNC. These findings expand the spectrum of titin’s roles in cardiomyopathies and provide novel insight into the molecular basis of titin-truncating variants-associated LVNC.
Left ventricular noncompaction (LVNC) is a relatively rare cardiomyopathy, with or without left ventricular dysfunction, characterized by excessively prominent trabeculations and associated deep recesses that communicate with the ventricular cavity.[1]LVNC is part of unclassified cardiomyopathies according to genetic cardiomyopathies by the America Heart Association. The prevalence of LVNC is estimated at 0.014% to 1.3%, depending on the age of patients.[2]The phenotypic expression and evolution of isolated LVNC are highly variable,and clinical features can range from asymptomatic to symptomatic, with a relatively stable course over several years or development towards severe complications, including congestive heart failure, ventricular arrhythmia and sudden cardiac death, atrial arrhythmias and systemic embolic events.[3-5]LVNC is supposed to be related to a premature arrest of compaction of the loose myocardial meshwork during fetal embryogenesis, with persistent trabeculated myocardium, but the precise pathophysiology remains poorly understood. The predominant mode of inheritance is autosomal dominant, with some cases with an X-linked transmission. Several genes have been identified as LVNC disease-causing.The first reported genetic cause of isolated LVNC was the X-linked tafazzin gene, which is also responsible for Barth syndrome.[6]The sarcomere-encoding genes (MYH7,ACTC1,TNNT2,MYBPC3,TMP1,andTNNI3) account for 17% to 30% of LVNC.[7,8]Other genes such as dystrobrevin alpha,Nkx-2.5, Z-line protein-encodingZASP/LDB3, lamin A/C (LMNA)genes have also been associated with LVNC.[9]Recently, several studies showed the importance of the titin gene (TTN) mutations in LVNC, as high as 19%in a German study of 68 index cases, 7% in adults from a Dutch cohort of 327 patients, and 10.5% in a French study of 95 unrelated adult patients.[4,10,11]
Titin is the largest human protein and spans half of the sarcomere. It contains four functionally distinct segments: (1) an N-terminus that is anchored at the Z disk; (2) a distensible I band; (3) an inextensible, thick filament binding A band; and (4) a carboxyl M band with a kinase domain. Titin-truncating variants (TTNtv) are associated with a highly variable phenotype.TTNtvis the most prevalent genetic cause of dilated cardiomyopathy, found in approximately 25% of familial cases.[12]TTNtvis also present in the general population, initially estimated at 1% to 2%.[13,14]Both TTN’s size and incomplete knowledge of the protein’s function in cardiomyocyte biology hindered the elucidation of why someTTNmutations produce clinical phenotypes.
The present study identified the presence of novelTTNtvp. R2021X mutation by whole-genome sequencing (WGS) in two family members, filtering against variants seen in normal population cohorts,and using linkage information derived from single nucleotide polymorphism arrays of four family members, and further explored the role and potential mechanism of the novelTTNtvp. R2021X mutationin vitrousing the clustered regularly interspaced short palindromic repeats associated protein 9 (CRISPR/Cas9) technology. These findings may represent a noteworthy contribution to clinical LVNC diagnostics and, potentially, to therapeutics.
METHODS AND MATERIALS
Clinical Evaluation
The study was approved by the Ethics Committee of Fuwai Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing,China (No.2021-1428), and all subjects provided written informed consent. A 3-generational family with a history of cardiomyopathy was recruited. Clinical assessment and genetic studies were performed on available family members, with clinical examination,electrocardiogram, and echocardiography. The diagnosis of LVNC was based on published criteria from echocardiographic or cardiac magnetic resonance imaging[15,16]: the compaction ratio, that is, the ratio of the thickness of noncompacted to compacted myocardium > 2.3 measured on magnetic resonance imaging in diastole or > 2.0 on echocardiography in systole, was used to diagnose LVNC.
Genetic Screening
Genomic DNA was extracted from peripheral blood leukocytes using the QIAamp DNA Blood Midi Kit (QIAGEN, Hilden, Germany), according to the standard protocols. The proband was selected for whole-exome sequencing. The Agilent SureSelect Human All Exon V5 capture kit (Agilent, Santa Clara,CA, USA) was used for exome capture. And the Illumina’s HiSeq4000 platform (Illumina, Inc, San Diego,CA, USA) was used for sequencing. Sequence data were aligned to the human reference genome (GRCh37/hg19) with BWA followed by sorting and marking of duplicate reads using Picard (version 2.4, http://broadinstitute.github.io/picard/). Local realignment of insertions/deletions (InDels) and base quality score recalibration were performed using GATK (version 3.6, https://www.broadinstitute.org/gatk/). GATK was also used to call and filter variants within a genome-wide region. The resultant variants were annotated with ANNOVAR[17]and sequentially filtered using the following criteria: (1) variants with an East Asian minor allele frequency ≥ 0.01 in the databases for 1000 Genomes Project (http://browser.1000genomes.org) and Exome Aggregation Consortium (ExAC, http://exac.broadinstitute.org) were removed; (2) variants in exons and splicing sites were retained; (3) certain types of variants were retained,including nonsynonymous, frameshift, nonframeshift insertion/deletion, stopgain and unknown;(4) variants in genes expressed in the heart according to the Human Protein Atlas (http://www.proteinatlas.org);[18]and (5) variants that passed manual confirmation using the Integrative Genomics Viewer(http://software.broadinstitute.org/software/igv)were retained. Effects of variants on splicing signals were evaluated using the Human Splicing Finder (version 3.0, http://www.umd.be/HSF3/HSF.html). Candidate pathogenic (likely) variants were validated by Sanger sequencing, performed with the primers: GCCTGAAGAGTCGGAAGAGC (Forward) and TTGCCTTCTTCGGCAAGAGC (Reverse).
CRISPR/Cas9 Gene Editing
A CRISPR/Cas9-mediated TTN R2021X mutation cell line was constructed. CRISPR/Cas9 H9C2 TTN R2021X mutation cell line was generated by designing sgRNAs using the web tools.[19]
Two pairs of sgRNA were annealed and cloned in PX459 vector at theBsmbI site according to GecKOZhang’s Lab Lentiviral CRISPR Tool Box [supplemental material, Figure 1S(A)]. The following primers were used for inserting sgRNAs into PX459 vector: primer 1: GGAGCTGAAGTCTCGCAAGA(Forward) and CCTTCGACTTCAGAGCGTTCT(Reverse); primer 2: CTTGCGAGACTTCAGCTCCA(Forward) and GAACGCTCTGAAGTCGAGGT (Reverse). Plasmids were purified from ampicillin-resistant colonies and sequence confirmed by Sanger sequencing using U6F primer/probe [supplemental material, Figure 1S(B)]. Then PX459-sgRNA were transduced in H9C2 cell line using a standard transduction protocol. H9C2 cells were sorted, expanded,and Sanger sequenced for genotyping [supplemental material, Figure 1S(C)].
Quantitative Reverse Transcription-polymerase Chain Reaction Analysis
Total RNA of wild-type (WT) and mutant cells was extracted using the Cell/Tissue Total RNA Isolation Mini Kit (Vazyme Biotech) according to the manufacturer’s instructions. A total of 2 ng of total RNA was reverse transcribed into complementary DNA using the HiScript II 1st Strand complementary DNA Synthesis Kit (Vazyme Biotech). Quantitative polymerase chain reaction (PCR) analysis was performed on a Real-Time PCR System (BIOER,Hangzhou, China) using ChamQ SYBR Master Mix(Vazyme Biotech). Primers are shown as following:TTN Z-disk: TCTCTCATCTCAGCCTCGGT (Forward) and TCTCTCATCTCAGCCTCGGT (Reverse); TTN M-line: AGGAACTCCTCCTCCCCATC(Forward) and CTCGTGCTGGTTTCTTCCCT (Reverse); and HPRT: CCCAGCGTCGTGATTAGTGATG(Forward) and TTCAGTCCTGTCCATAATCAGTCC(Reverse). Each sample was performed in triplicate.HPRT served as an internal reference. The thermo cycle conditions were set as follows: denaturation at 95 °C for 30 s, followed by 40 cycles of denaturation at 95 °C for 10 s and extension at 60 °C for 30 s. Data were analyzed using the 2-ΔΔCTmethod.
Western Blot Analysis
Changes in protein expression levels in WT and mutant cells were determined by Western blot analysis. The M-PER Mammalian Protein Extraction Reagent (ThermoFisher Scientific) was applied to extract the protein. A total of 20 μg of the protein was separated using 10% sodium dodecyl sulfatepolyacrylamide gel electrophoresis at 80 V for 100 min. Then, proteins were transferred onto polyvinylidene difluoride membranes (Millipore, Burlington, MA, USA) using 300 mA for 180 min. Membranes were blocked with 5% non-fat milk dissolved in Tris-buffered saline with 0.1% Tween-20 (TBST)for 1 h at room temperature (RT), followed by incubation with the primary antibodies. Subsequently, membranes were washed three times in TBST for 5 min, followed by incubation with horseradish peroxidase-conjugated goat anti-rabbit IgG secondary antibody (1:10000, EarthOx Life Sciences, Millbrae, CA, USA) for 1 h at RT, then washed three times in TBST. Finally, proteins were visualized by chemiluminescent detection using an emission chemoluminescence detection kit (ThermoFisher Scientific).Images and the density of each band were acquired on a Fusion-FX6 imaging system (Vilber Lourmat).To compare the expression of proteins, HPRT served as an internal control.
Cellular Immunofluorescence
WT and mutant cells were fixed in 4% paraformaldehyde solution, permeabilized with 0.25% Triton X-100, blocked in 3% bovine serum albumin,and incubated with the primary antibody (mouse anti-titin 1:50, Santa Cruz 271946) overnight at 4 °C.Cells were then washed with phosphate buffered saline (PBS) and incubated with secondary antibody(goat anti-mouse conjugated with Alexa Fluor 546,1:1000, Invitrogen) at RT for 2 h. The cells were then washed with PBS, fluorescence images were acquired and fluorescence intensity was detected using the laser confocal scanning microscope (Zeiss LSM710).
Extracellular Oxygen Consumption Assay
Oxygen consumption assay was performed using the Extracellular Oxygen Consumption Assay(Abcam, ab197243) following the manufacturer’s protocol. Plates were analyzed by Spark 20M multimode microplate reader (Tecan Group Ltd., Mannedorf, Switzerland) using time-resolved fluorescence.Signals were read every 90 s for 120 repeats with an optimal delay time of 30 μs and gate (integration) time of 100 μs. The signal from wells without cells was used as a background signal.
Measurement of Adenosine Triphosphate (ATP)Levels
We used a luminescent ATP detection assay kit(Abcam, ab113849) with Spark 20M multimode microplate reader (Tecan Group Ltd., Mannedorf, Switzerland) to measure ATP levels. A total of 1 × 105WT and mutant cells were cultured in 96-well cell culture plates 24 h prior to the assay. The process was performed according to the manufacturer’s instructions.
ETC Complex Activity Assays
Mitochondrial respiratory chain activities involving complex I (ab109721), II (ab109908), IV (ab10 9911), and V (ab109714) were measured using kits from Mitoscience-Abcam according to the manufacturers’ instructions. Briefly, capture antibodies specific for each mitochondrial respiratory chain complex were precoated in each well of a 96-well microplate. Cell cultures or isolated mitochondria were added to the microplate wells, which had been precoated with a specific capture antibody. After the target had been immobilized in the well, the complex activity was determined by following the oxidation and reduction of complex-specific substrates and a dye. The absorbance is measured at specific optical densities (OD) using a spectrophotometer.
Complex I activity was determined by following the oxidation of NADH to NAD+and the simultaneous reduction of a dye which leads to increased absorbance at OD = 450 nm.Complex II activity was determined by the production of ubiquinol coupled to the reduction of the dye DCPIP (2,6-diclorophenolindophenol) and a decreases in its absorbance at 600 nm. Complex IV activity was determined by the oxidation of reduced cytochrome c by the absorbance change at 550 nm. Complex V activity was measured by monitoring the decrease in absorbance at 340 nm. Complex III activity was measured using a kit from BioVision (K520) according to the manufacturer’s instructions. Briefly, this kit for complex III activity assay is based on the reduction of cytochrome c through the activity of complex III. The absorbance of reduced cytochrome c is measured at 550 nm in isolated mitochondria.
Measurement of Mitochondrial Membrane Potential
Mitochondrial membrane potential was detected using the JC-1 assay (Dojindo, MT 09) according to the manufacturer’s protocol. The principle of the detection of JC-1: JC-1 dye showed potential-dependent accumulation in mitochondria. In normal mitochondria, JC-1 aggregates in the mitochondrial matrix to form polymers, which emit strong red fluorescence (Ex = 585 nm, Em = 590 nm). Unhealthy mitochondria due to the decrease or loss of membrane potential, JC-1 can only exist in the cytoplasm as a monomer and produce green fluorescence (Ex = 514 nm,Em = 529 nm). So the change in color is a very direct reflection of the change in mitochondrial membrane potential. The degree of mitochondrial depolarization can also be measured by the ratio of red to green fluorescence intensity. Cells were washed with PBS and were then stained with JC-1 probe for 30 min at 37 C/5% CO2in the dark. Subsequently, The red-to-green fluorescence ratio was employed to evaluate the changes in mitochondrial membrane potential.
Electron Microscopy
Cells grown on glass coverslips were fixed with 4% paraformaldehyde and 2.5% glutaraldehyde in PBS, dehydrated in a graded series of ethanol, and embedded in Epon. After removing the coverslip,ultrathin sections were cut on a Reichert Ultracut microtome, positively stained with aqueous uranyl acetate and lead citrate and viewed in a Philips CM 10 electron microscope at an accelerating voltage of 80 kV. Pictures were taken with a slow-scan CCD camera (Gatan, Pleasanton, CA, USA).
Statistical Analysis
Data are presented as mean ± SD. Statistical analysis were performed using GraphPad Prism version 6.0 (GraphPad Inc., La Jolla, CA, USA). Comparisons were made using the Student’st-test or oneway analysis of variance followed by Tukey’s multiple comparison test. Two-sidedP-value < 0.05 were considered statistically significant.
RESULTS
Clinical Evaluation of the Pedigree
The proband was a 55-year-old male (II-1 in Figure 1A) who presented chest tightness and shortness of breath in daily activities. 24-hour Holter electrocardiogram monitoring suggested persistent atrial fibrillation and nonsustained ventricular tachycardia.Echocardiography suggested LVNC with left ventricular dilatation and severe systolic dysfunction (Figure 1B; left ventricular end diastolic dimension = 75 mm,left ventricular ejection fraction = 28%). His father (I-1)died of decompensated heart failure at the age of 60 years. Cascade screening identified that his son (III-1)had sufficient noncompaction to meet the diagnostic criteria for LVNC, with frequent ventricular premature beats accompanied with normal diastolic and systolic function. The proband’s mother was found to have both normal heart structure and function.
Identification of TTN p. R2021X Segregating With Disease
The proband and his son were selected for WGS that identified a heterozygous single-base alteration at position 6061 (c.6061C > T) inTTN, the mutation site is located in the exon area, which resulted in a premature stop of titin protein (p. R2021X) (supplemental material, Figure 1S).
This mutation has not been identified in the Ex-AC database. Andin silicoprediction (Mutation Taster) reported a deleterious effect. Sanger sequencing confirmed the cosegregation of the heterozygous mutation with disease in all affected individuals of the family (Figure 1C). Thus, comprehensive whole-genome analysis with reveals the p. R2021X mutation inTTNas the most plausible causative mutation in the family.
Functional Studies in TTN R2021X Mutation Model
Nonsense-mediated mRNA decay and sarcomere deficits caused LVNC phenotypeTo quantify cardiac titin expression after mutation ofTTNR2021X using the sgRNA2 guided CRISPR/Cas9 system, we performed real-time RT-PCR analysis with oligonucleotides specific for titin’s Z-disc portion (amplifying both alleles), or with allele-specific oligonucleotides(amplifying only the wild-type allele of titin’s M-line portion). Results showed thatTTNmutant allele was degraded in mRNA level in the mutant cells (Figure 2A & 2B). To detect titin protein level, we performed immunofluorescence using anti-titin antibodies with binding sites C-terminal of titin. A minimal amount of approximately 1% titin protein was observed in the mutant cells, suggesting a haploinsufficient disease mechanism in the presence of theTTNR2021X mutation (Figure 2C).
TTN R2021X mutation led to mitochondrial dys-functionThe mitochondrial respiratory activity represented by cellular oxygen consumption rate(OCR) and ATP level was measured in WT andTTNR2021X mutant cells. Mutant cells showed a significant reduction of OCR (Figure 3A,P< 0.001) and ATP level (Figure 3B,P< 0.001), indicating that the decreased expression of full-length titin led to impaired mitochondrial respiration. Activities of the electron transport chain complexes (ETCs), involving complex I to IV as well as the ATP synthase (complex V), were then measured. Compared to WT cells,TTNR2021X mutant cells showed reduced activity of complex I and compex V (Figure 4C and Figure 4,P< 0.01 andP< 0.001), while there was no significant difference of other ETCs between the two groups(Figure 3D-3F).

Figure 1 Clinical and genetic profile of the pedigree. (A): Pedigree of the family; males depicted as squares; females, circles; slanted symbols, deceased individuals. Clinically affected individuals are marked in black, unaffected are shown in white; (B): echocardiogram images showing the characteristic spongy appearance of noncompaction ( indicated by yellow arrows) in individual II-1. (a) long axis section of left ventricle; (b) apical four chamber section; (c) short axis section of left ventricle; (d) M mode (left ventricular end diastolic diameter indicated by red line, left ventricular end systolic diameter indicated by blue line); and (C): Sanger sequencing chromatogram showing a heterozygous c.C6061 > TTN p. R2021X truncating mutation in TTN gene.
The changes of membrane potential of mitochondria were also qualified, illustrated with the significant decrease of the fluorescence intensity of JC-1 inTTNR2021X mutant cells compared with WT cells,indicating mitochondrial dysfunction (Figure 4A,P< 0.001). Damaged mitochondria often exhibit structural changes, including formation of giant mitochondria (megamitochondria), fragmentation of mitochondria, or other shape changes. Indeed, we observed abnormal mitochondrial structures inTTNR2021X mutant cells under electron microscopy (Figure 4B). Taken together, the co-occurrences of impaired mitochondrial respiratory activity, reduced mitochondrial membrane potential, and structural changes of mitochondria suggested the accumulation of dysfunctional/damaged mitochondria induced byTTNR2021X mutation.

Figure 2 Nonsense-mediated mRNA decay and haploinsufficiency mechanism in TTNtv p. R2021X mutant cells. (A): RT-PCR showing titin mRNA level (n = 3); and (B): cellular immunofluorescence showing titin protein level (n = 3).
Impaired autophagy and activated Akt- mTORC 1 signaling pathway in mutant cellAutophagyrelated ultrastructural differences between WT andTTNR2021X mutation cells were evaluated under electron microscopy. We observed markedly fewer numbers of active lysosomes in the mutant cells than WT cells, suggesting impaired autophagy induced byTTNR2021X mutation (Figure 5). Furthermore,an accumulation of glycogen granules was identified inTTNR2021X mutation cells, which indicated the impaired autophagic pathway initiated byTTNR2021X mutation.
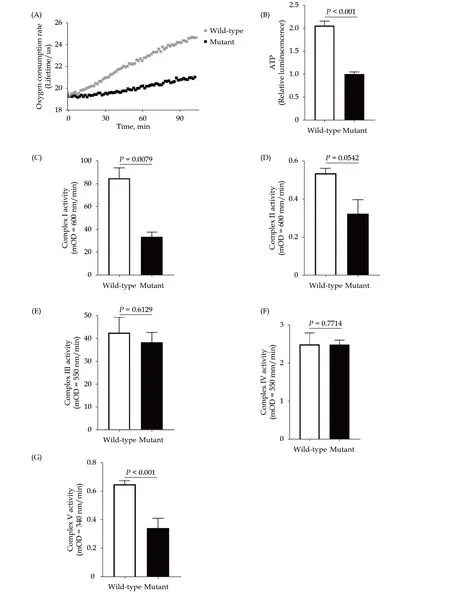
Figure 3 Impaired mitochondrial oxidative phosphorylation capacity in TTNtv p. R2021X mutant cells. (A): Oxygen consumption rate; (B): ATP level; (C): activity of mitochondrial complex I; (D): activity of mitochondrial complex II; (E): activity of mitochondrial complex III; (F): activity of mitochondrial complex IV; and (G): activity of mitochondrial complex V.
Mammalian target of rapamycin complex 1 (mTORC1) is a key negative regulator of autophagy. Hence,we evaluated mTORC1 signaling. Levels of phosphorylated proteins of mTOR, ribosomal protein S6,and eukaryotic translation initiation factor 4E-binding protein 1 were higher inTTNR2021X mutation cells than WT cells (Figure 6A & 6B, Figure 6E and Figure 6H,P< 0.001 ), together with the decreased expression levels of total mTOR, S6, and 4E-binding protein 1 in the mutant cells compared with WT cells(Figure 6A, Figure 6C, Figure 6F and Figure 6I,P<0.001). Then the ratio of phosphorylated-to-total proteins was increased in the mutant cells compared with WT cells (Figure 6D, Figure 6G and Figure 6J,P< 0.001), suggesting an activated mTORC1 signaling transduction caused byTTNR2021X mutation.In addition, the dual phosphorylation of serine-threonine kinase Akt/protein kinase B, demonstrated by p-Akt (Thr308)/Akt and p-Akt (Ser473)/Akt ratios, was significantly increased inTTNR2021X mutation cells compared with WT cells (Figure 6K-6O,P< 0.001). Combined, these results suggested that activated mTORC1 might result from the activation of Akt signaling in response toTTNR2021X mutation.

Figure 4 Dysfunctional/damaged mitochondria in TTNtv p. R2021X mutant cells. (A): Representative pictures of mitochondrial membrane potential in wild-type and mutant cell; (B): quantification of red/green fluroscence intensity; and (C): mitochondrial structure under electron microscopy. (a) Overview of mitochondrial morphology and distribution in wild-type cells (× 5000 magnification); (b) normal mitochondrial morphology and structure in wild-type cells (× 10,000 magnification); (c) normal mitochondrial morphology and structure in wild-type cells (× 10,000 magnification); (d) overview of mitochondrial morphology and distribution in mutant cells (× 10,000 magnification); (e) mitochondrial demyelination degeneration in mutant cells (× 10,000 magnification, indicated by black arrows); and (f) mitochondrial membrane damage in mutant cells (× 10,000 magnification, indicated by black arrows).
DISCUSSION
In the present study, we found a novel mutation inTTNamong highly penetrant familial LVNC patients. The three-generation family with three individuals affected by LVNC was presented with clinical features of arrhythmia and systolic impairment and an autosomal dominant inheritance pattern. A combination of WGS and linkage analysis identifiedTTNp. R2021X mutation as a genetic cause. Further functional studiesin vitrosuggested a potential pathogenic role ofTTNp. R2021X, in whichTTNp. R2021X mutation led to titin mRNA degradation and titin protein haploinsufficiency, mitochondrial dysfunction indicated by impaired mitochondrial respiratory activity, reduced mitochondrial membrane potential, and abnormal ultrastructural changes, and decreased autophagy associated with activated mTORC1 signaling pathway.
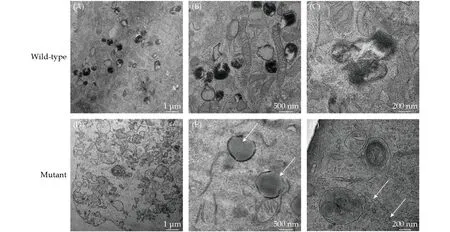
Figure 5 Electron microscope images showing impaired autophagy in TTNtv p. R2021X mutant cells. (A-C): Normal number of active lysosomes in wild-type cells; (D): less number of active lysosomes in mutant cells; (E): accumulation of lipids in mutant cells (indicated by black arrows); and (F): accumulation of glycogen granula in mutant cells ( indicated by white arrows).
To our knowledge, this is a novelTTNtruncating mutation implicated in LVNC. LVNC is genetically heterogeneous and can be inherited as an autosomal dominant or X-linked recessive disorder. It has been linked to mutations in many genes, including LIM domain-binding protein 3 (ZASP), a-dystrobrevin(DTNA), tafazzin (TAZ/G4.5), and those encoding sarcomeric, Z-disc, cytoskeleton proteins, and mitochondria.[6,7,20]Recently, several studies showed the importance ofTTNgene mutations in LVNC. A missense mutation inTTNwas first reported as the cause of a highly penetrant familial cardiomyopathy with features of LVNC in 2016.[10]An Dutch multicenter study analyzed 327 unrelated patients with LVNC and showed that mutations inTTNfrequently occurred in adults (7%,n= 18).[11]Further study proposed thatTTNwas the most prevalent disease gene of LVNC, with a high mutation rate of 19%.[21]In contrast to the recent broad TTNtv mutation research, our finding ofTTNp. R2021X as a genetic cause in a LVNC family expands the spectrum of titinopathies.
Haploinsufficiency is now generally accepted to be the molecular mechanism forTTNtv. This means that transcripts ofTTNtvmay undergo nonsensemediated decay, so that the abnormal protein is never expressed, leading to a reduced protein dose.
More recent work showed that DCM causingTTNtvis enriched in the sarcomeric A-band region; however,I-bandTTNtvhas been identified in healthy individuals and the general population without DCM.[13]In previous studies of cardiomyocytes derived from induced pluripotent stem cells from DCM patients withTTNtv, or created using CRISPR/Cas9 gene-editing technology, functional and RNAseq data suggest alternative exon splicing as the predominant mechanism for reduced penetrance of I-bandTTNtv.[22]TTNtvhave also been associated with various other clinical phenotypes, including peripartum cardiomyopathy, arrhythmogenic right ventricular cardiomyopathy, hypertrophic cardiomyopathy, restrictive cardiomyopathy, and a range of skeletal myopathies,[12,23-25]In this study, RT-PCR and cell immunofluorescence data supported a haploinsufficient disease mechanism inTTNp. R2021X mutation. However,it is still unclear how this mutation leads to this distinct phenotype; more insight into the genetic susceptibilities or an environmental modifier ofTTNgene expression is needed to understand the underlying signaling pathways in LVNC.

Figure 6 Increased Akt-mTORC1 signaling in TTNtv p. R2021X mutant cells. Protein levels of p-mTOR, mTOR, p-S6, S6, p-4EBP1, 4E-BP1, phosphorylated Akt ( p-AktThr308, p-AktSer473) and Akt in wild-type and mutant cells. (A): Representative Western blots of indicated proteins. HPRT was used as loading control; and (B-O): quantification of p-mTOR, p-mTOR, p-mTOR/mTOR, p-S6,S6, p-S6/S6, p-4E-BP1, 4E-BP1, p-4E-BP1/4E-BP1, p-AktSer308, p-AktSer473, Akt, p-AktThr308/Akt, p-AktSer473/Akt.
Here, we provided several lines of evidence that demonstrated mitochondrial dysfunction in the presence ofTTNp. R2021X mutation. Remarkable reduction of OCR, ATP level, and ETCs activity indicated impaired mitochondrial respiratory activity in the mutant cells. Indeed, abnormal mitochondrial structures were identified in the mutant cells under electron microscopy. Our findings are in line with the metabolic remodelling results found in bothTTNtv-positive DCM patients andTTNtvmutated rat hearts model.[13,26,27]Similar to our findings,TTNtv-positive DCM patients demonstrated altered mitochondrial energetics comparing withTTNtvnegative DCM patients.[26]TTNtv-positive DCM rat hearts also showed mitochondrial abnormalities and metabolic shift compared to wild type counterpart. Metabolic shift from fatty acid oxidation towards glycolysis was observed inTTNtvanimal models.[27]It is suggested that the metabolic shift inTTNtvhearts may be a maladaptive response from mitochondrial dysfunction.TTNtvhearts are in a compensated state trying to counterbalance aTTNtv-related sarcomeric defect.[13,22]
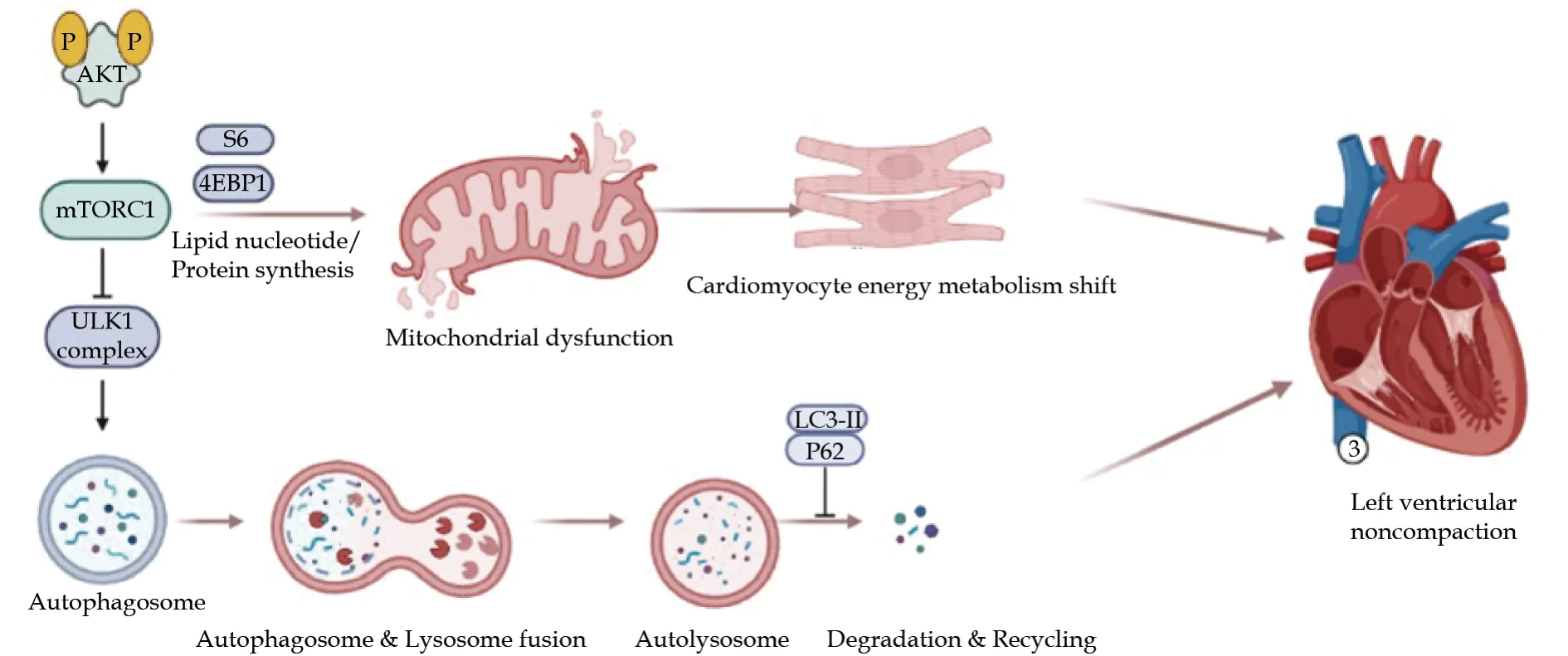
Figure 7 TTNtv p. R2021X mutation and left ventricular noncompaction. TTNtv p. R2021X mutation cause mitochondrial dysfunction and decreased autophagy associated with activated Akt/mTORC1 signaling pathways in cardiomyocytes, leding to left ventricular noncompaction phenotype.
However, to date, the molecular mechanism(s) underlying the mitochondrial dysfunctions inTTNtv-associated cardiomyopathy is not clear yet.
Emerging evidence has suggested an essential role for basal autophagy in maintaining normal cardiac function.[28]Activation of mTORC1 and decreased autophagy has also been observed in several cardiomyopathy models and can be rescued by mTORC1 inhibition.[29-31]Furthermore, defective autophagy led by activation of mTORC1 signaling in theTTNtvDCM rat hearts has been demonstrated.[27]
In present stusy, we observed markedly fewer numbers of active lysosomes in the mutant cells than WT cells, suggesting impaired autophagy induced byTTNR2021X mutation. Evidence of impaired autophagy was identified inTTNp. R2021X mutation cells associated with the activated mTORC1 signaling (Figure 7). However, our result is different from previous studies. According to previous studies, accumulation of p62 protein and autophagosomes were observed inTTNtvhearts, which indicated defective lysosomal degradation rather than decreased autophagosome formation. However,the exact mechanism still needs further investigation to clarify the mTOR signaling in the molecular mechanism of lysosome function. In our study, p-Akt (Thr308)/Akt and p-Akt (Ser473)/Akt ratios were significantly increased inTTNR2021X mutation cells compared with WT cells, demonstrating that Akt signaling was activated. Our result suggested that activated mTORC1 might result from the activation of Akt signaling in response toTTNR2021X mutation. However, the exact mechanism of how Akt signaling results in mTORC1 activation is still unclear. Further studies needs to be done to clarify the exact mechanism. According to a previous study in Mybpc3-targeted knockin mice Akt/PKB signaling contributing to mTORC1 activation was associated with the upregulation of Ctgf, encoding the connective tissue growth factor, which induced cardiomyocyte hypertrophy via Akt signaling.[30]
LIMITATIONS
Several limitations still exist in the present study.The novelTTNtvmutation was identified in a small-sized LVNC pedigree with limited clinical samples;hence, a large clinical cohort needs to be established to determine the pathogenicity ofTTNp. R2021X mutation in LVNC and clarify the association between particular genotypes and distinct phenotypes in future work. Secondly, functional analysis was only carried in the CRISPR/Cas9-mediatedTTNR2021X mutationin vitro; further available studies on animal models and patient-derived induced pluripotent stem cell-derived cardiomyocytes are needed to explore the mechanistic link betweenTTNtvand mTOR signaling.
CONCLUSIONS
In this study, we identified a novel truncating mutation inTTN,TTNp. R2021X, as a genetic cause of LVNC, which expands the spectrum of titinopathies. To our knowledge, this is the firstTTNtvimplicated in LVNC supported by robust genome-wide genetics and detailed functional data, that is,TTNp. R2021X mutation led to titin mRNA degradation and titin protein haploinsufficiency, mitochondrial dysfunction, and decreased autophagy associated with activated Akt/mTORC1 signaling pathways in cardiomyocytes. These findings provide novel insight into the molecular basis and identify several potential novel drug targets for clinical intervention ofTTNtv-positive LVNC patients.
ACKNOWLEDGMENTS
This study was supported by the National Key Research and Development Program of China (2016 YFC1300100), the National Natural Science Foundation of China (No. 81974042), the Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences (2019XK320058) , and the Peking Union Medical College Youth Fund (No.3332018058). All authors had no conflicts of interest to disclose.
 Journal of Geriatric Cardiology2022年4期
Journal of Geriatric Cardiology2022年4期
- Journal of Geriatric Cardiology的其它文章
- Ambulatory diastolic blood pressure: a marker of comorbidity in elderly fit hypertensive individuals?
- Essen score in the prediction of cerebrovascular events compared with cardiovascular events after ischaemic stroke or transient ischaemic attack: a nationwide registry analysis
- Pre-hospital delay in patients with acute myocardial infarction in China: findings from the Improving Care for Cardiovascular Disease in China-Acute Coronary Syndrome (CCC-ACS) project
- Tongmai Yangxin Pill combined with metoprolol or metoprolol alone for the treatment of symptomatic premature ventricular complex: a multicenter, randomized, parallel-controlled clinical study
- Trends and sex differences in atrial fibrillation hospitalization and catheter ablation at tertiary hospitals in China from 2013 to 2016
- Invasive versus non-invasive hemodynamic monitoring of heart failure patients and their outcomes